空间转录组上游分析
转录组上游分析,即将原始的reads数据与人类参考基因组进行比对(明确每一条reads属于哪个基因),从而将reads数据转为counts数据。对于空间转录组,我们的目标是从原始的测序文件得到每一个测序spot中每个基因的表达丰度。
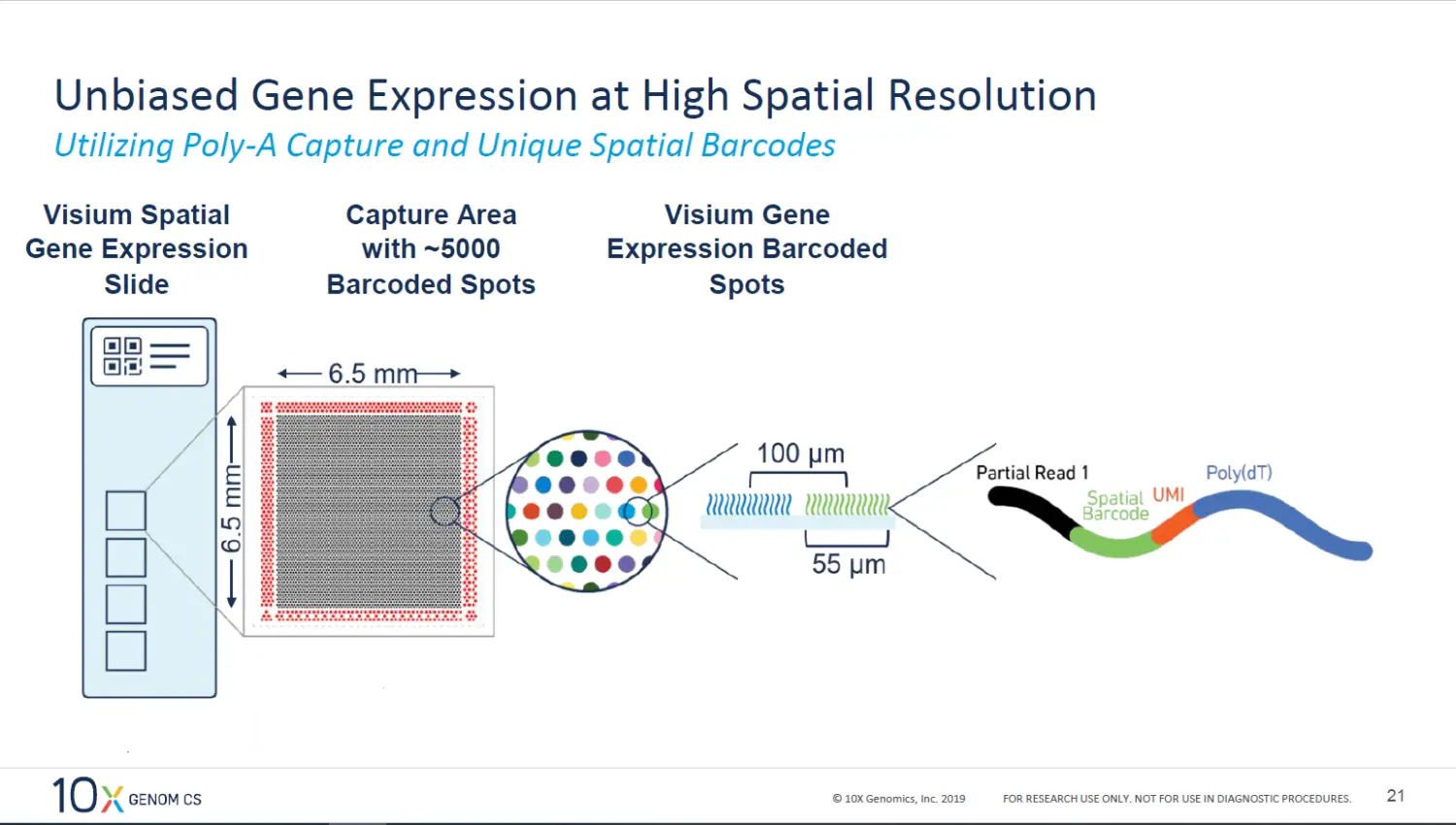
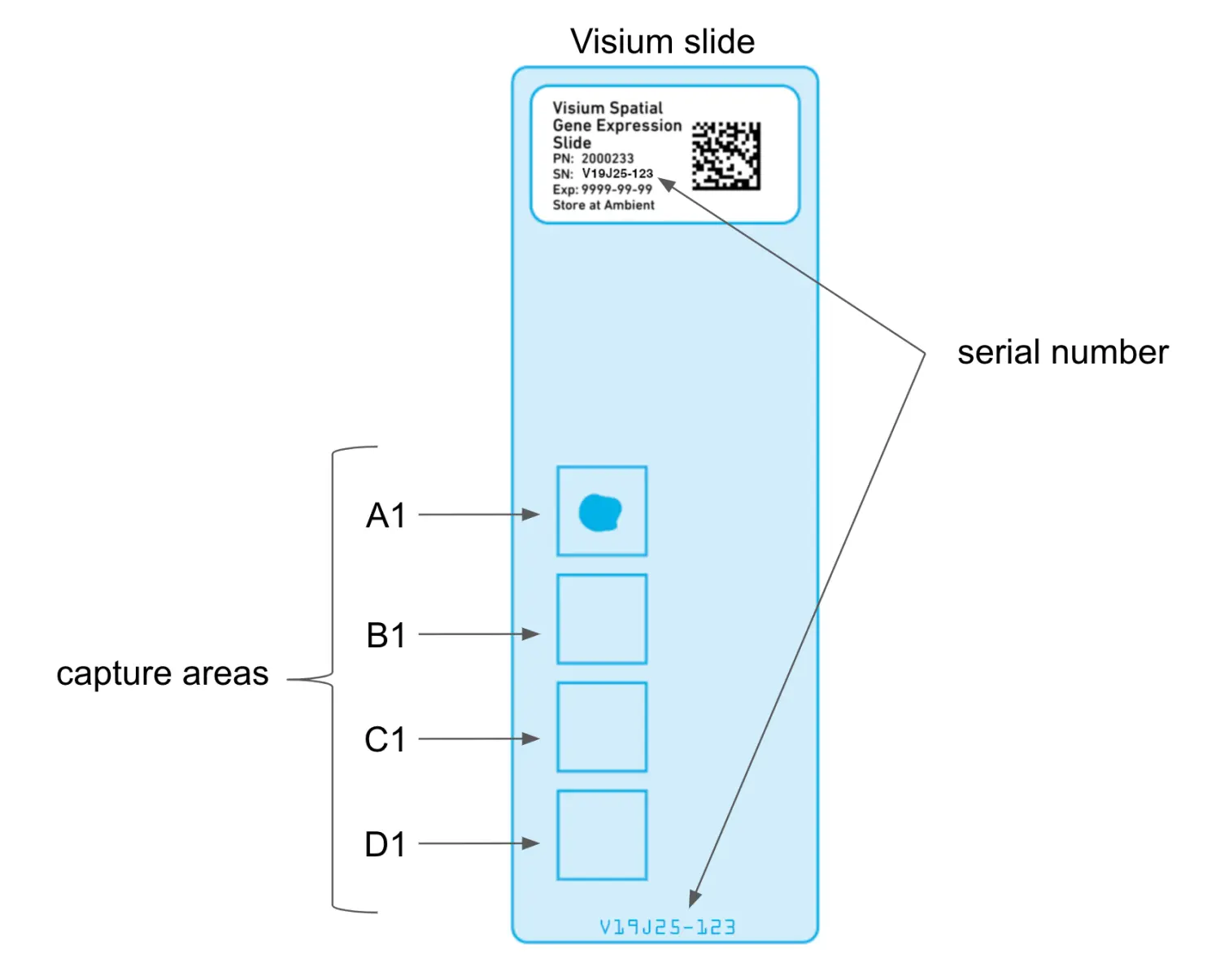
测序芯片的构成
每个测序芯片上都有四个区域,分别为A1,B1,C1,D1,并且还有一个唯一的序列号。在进行Space Ranger分析的时候,必须提供载玻片的序列号,以及与每个样本的捕获区域。官方的服务器上存储了每张序列号的测序芯片的layout信息,即每个spot的barcode序列以及对应的空间坐标,故运行spaceranger的时候需要连接互联网。
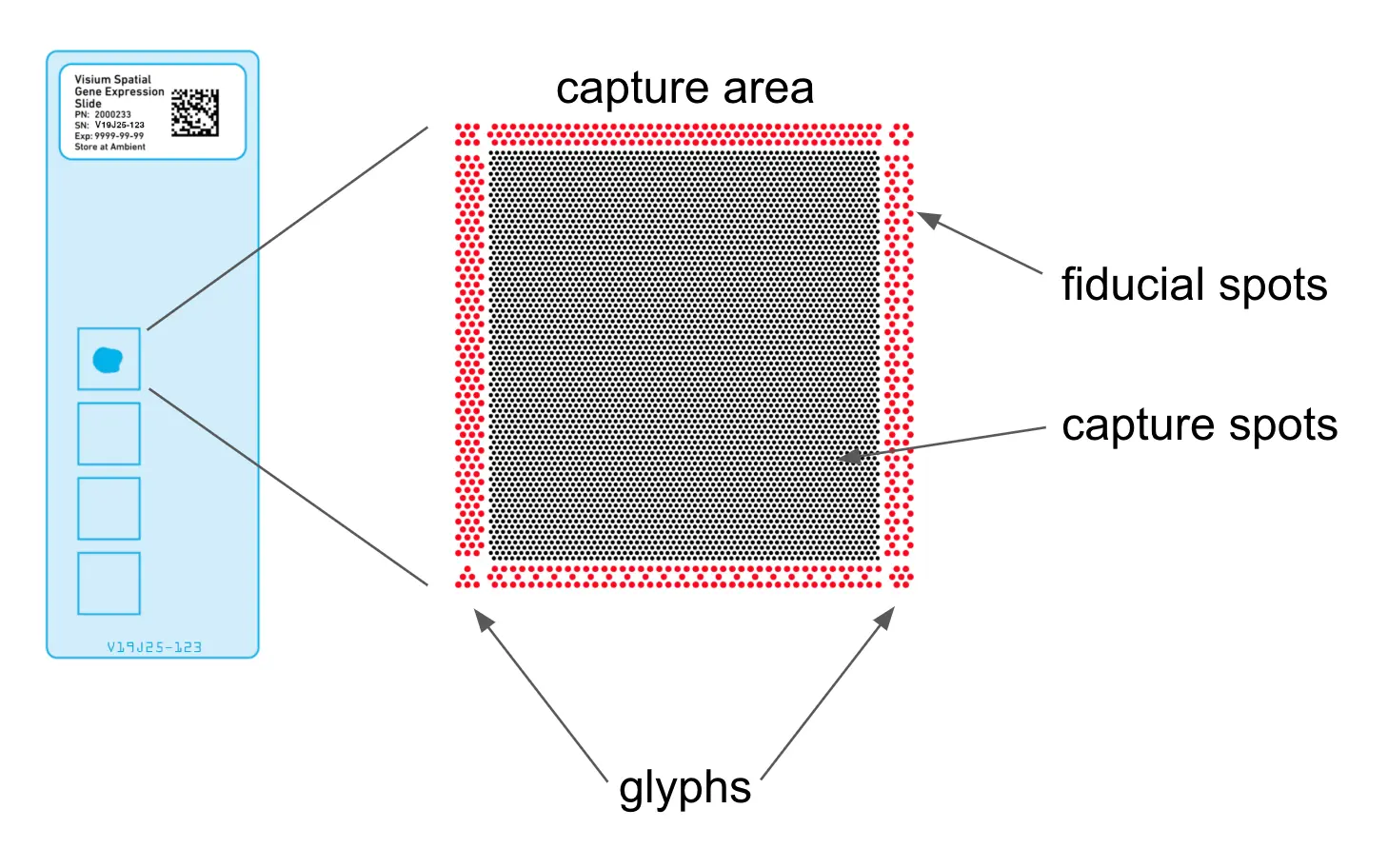
测序区域的构成
在每个测序区域包括以下部分:
capture spots:捕获位点,每个位点中都有众多寡核苷酸用于捕获mRNA,每一条寡核苷酸都由测序接头-空间Barcode-唯一分子标识符UMI-polydT四部分构成,其中每一个spot中barcode是一致的,而UMI是独一无二的。每个捕获点的直径约为55微米,大致可以容纳十个细胞。
fiducial spots:基准点,即围绕每个捕获区域的由圆形构成的规则图案。spaceranger会基于图像识别技术,根据这些基准点进行空间定位以明确每个capture spots在图像上的坐标。
glyphs:glyph标记,即捕获区域四角处的基准点,分别为沙漏型、开放六边形、三角形和填充六边形,用来区分上下左右。
数据与软件的准备
下载示例数据
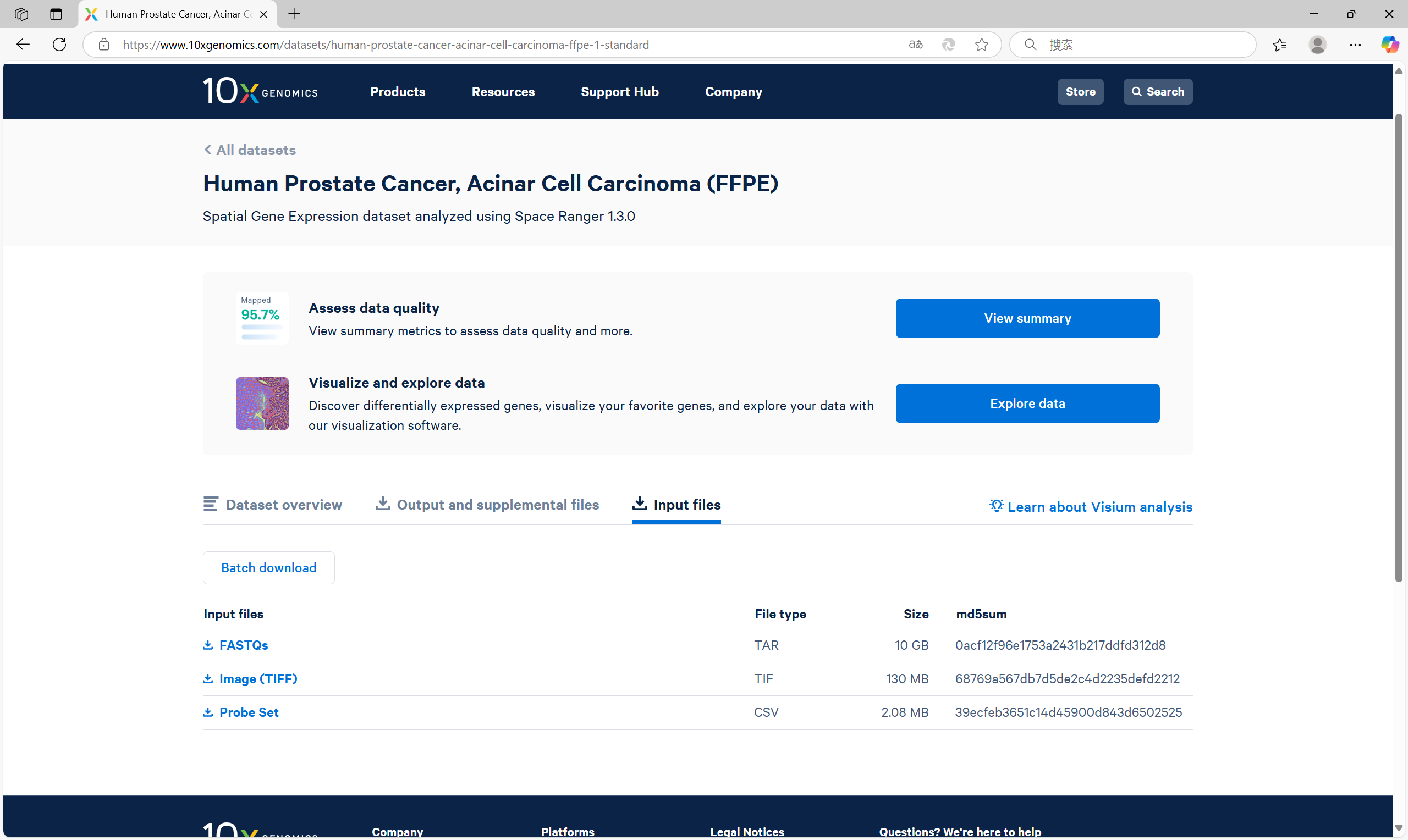
我们选用10XGenomics官网上的前列腺癌示例数据进行分析。这里需要下载三个文件:
FASTQs:包含测序读取的原始数据,用于基因表达分析。Image (TIFF):组织切片的数字图像。结合FASTQ文件中的测序数据,可以重建基因表达的空间分布图。Probe Set:使用的基因探针的集合。(记录了每个基因使用的捕获序列分别是什么)
值得注意的是,对于某些Probe Set,线粒体和核糖体基因以及许多经历体细胞重排的免疫基因被排除在外;编码序列小于 50 bp 的基因也被排除在外。故在某些样本的后续分析中是看不到线粒体基因的。
FASTQ文件
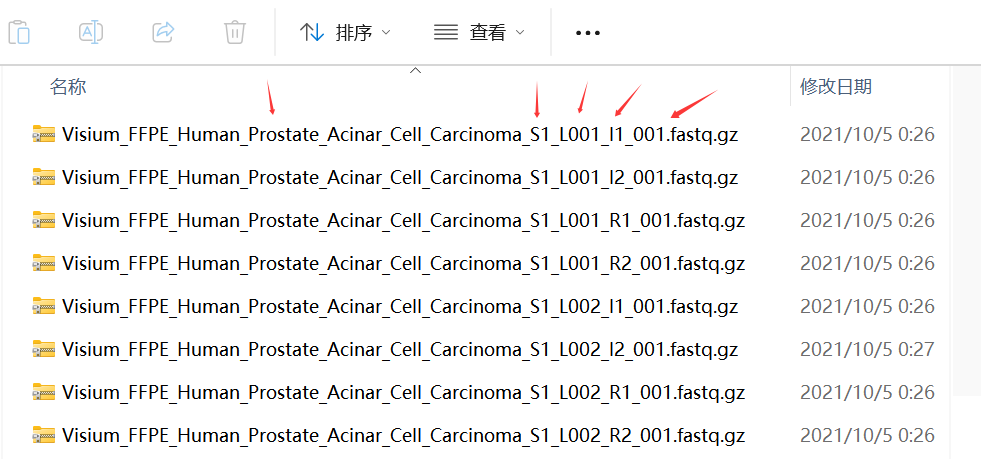
以第一个文件为例,其中Visium_FFPE_Human_Prostate_Acinar_Cell_Carcinoma为样本基本名;S1指样本编号;L001指测序时lane的编号;I1/I2和R1/R2分别指sample的index1/2,和Read1/2;001:每个测序文件均为001结尾,无特殊含义。
Image文件
包含了通过显微拍摄的样本的tiff格式照片

Probe Set文件
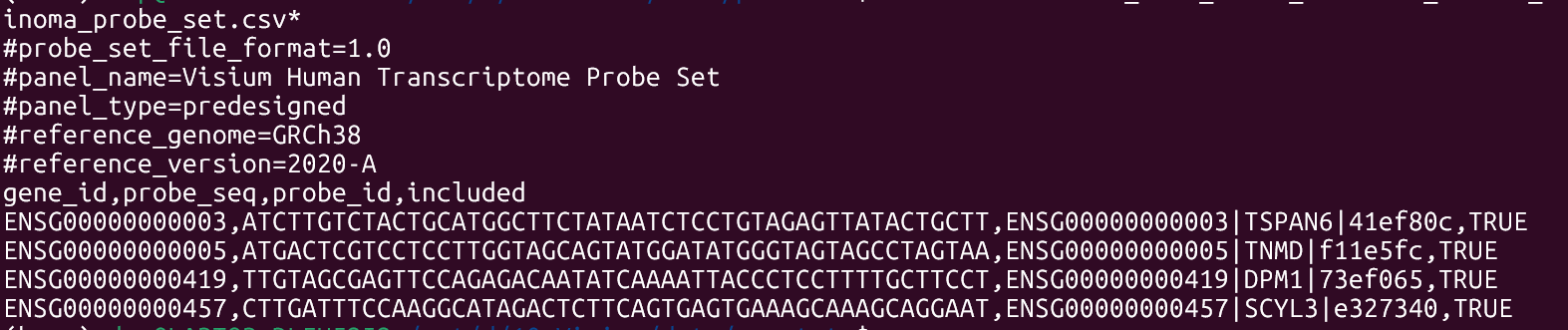
一个tsv格式文件,标记了每个基因用的是什么探针序列。
在官网上进行下载
1
2
3
4
5
6
7
8
|
wget -c https://cf.10xgenomics.com/samples/spatial-exp/1.3.0/Visium_FFPE_Human_Prostate_Acinar_Cell_Carcinoma/Visium_FFPE_Human_Prostate_Acinar_Cell_Carcinoma_fastqs.tar
wget -c https://cf.10xgenomics.com/samples/spatial-exp/1.3.0/Visium_FFPE_Human_Prostate_Acinar_Cell_Carcinoma/Visium_FFPE_Human_Prostate_Acinar_Cell_Carcinoma_image.tif
wget -c https://cf.10xgenomics.com/samples/spatial-exp/1.3.0/Visium_FFPE_Human_Prostate_Acinar_Cell_Carcinoma/Visium_FFPE_Human_Prostate_Acinar_Cell_Carcinoma_probe_set.csv
# 解压
tar -xvf Visium_FFPE_Human_Prostate_Acinar_Cell_Carcinoma_fastqs.tar
|
下载人类参考基因组
下载GRCh38参考基因组并解压:
1
2
|
wget -c https://cf.10xgenomics.com/supp/spatial-exp/refdata-gex-GRCh38-2020-A.tar.gz
tar -zxvf refdata-gex-GRCh38-2020-A.tar.gz
|
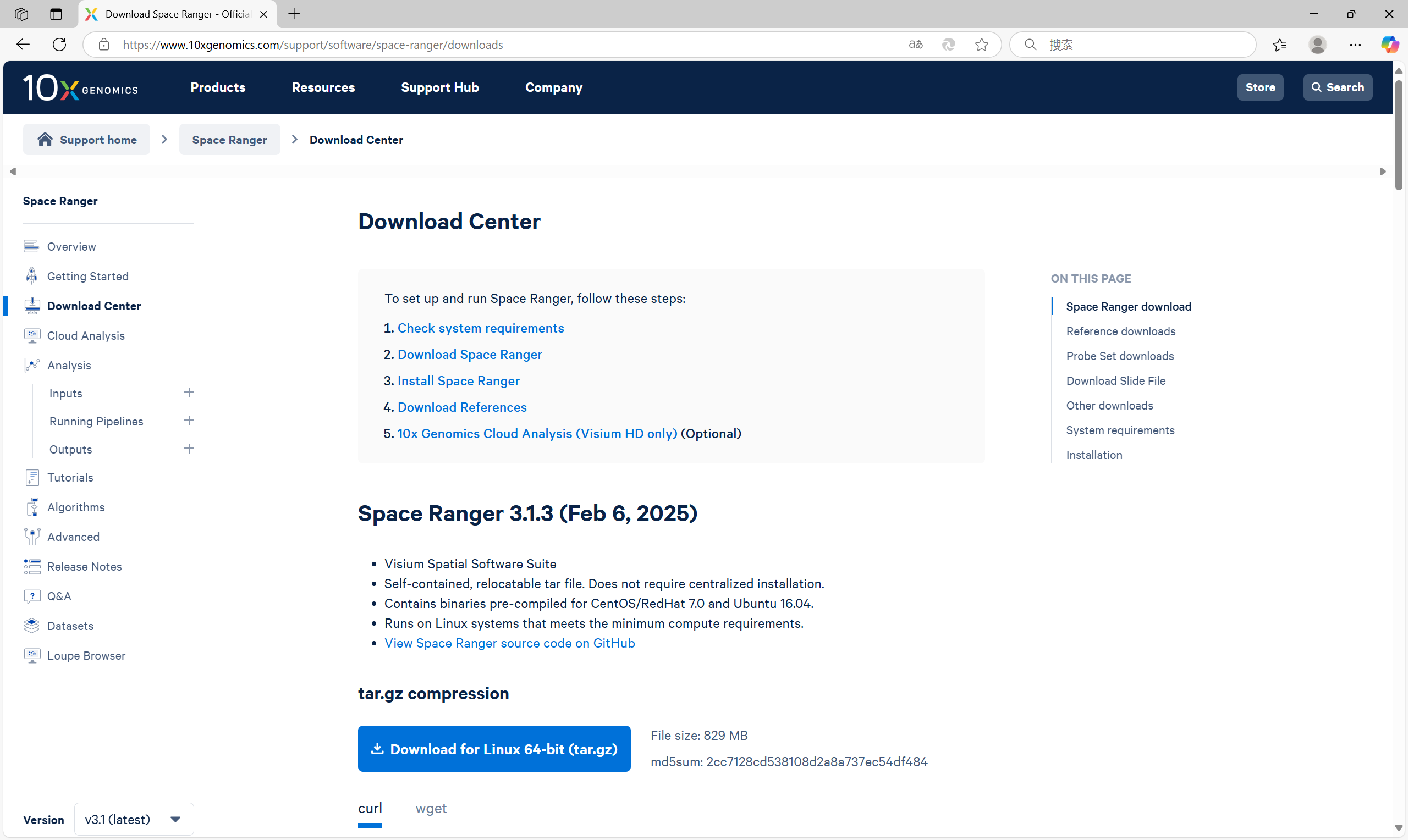
下载Space Ranger软件
1
2
3
4
5
6
7
8
|
# 下载spaceranger软件【version=3.1.3】
wget -O spaceranger-3.1.3.tar.gz "https://cf.10xgenomics.com/releases/spatial-exp/spaceranger-3.1.3.tar.gz?Expires=1743088491&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA&Signature=lXRXzqhcH1zlQ9QhvPqix8FJwbZSFTMAIWJggVNAZ-fw5lF9KqZOuTdQepBWejw9fsfN0608rxkia~vcQCQaUORoUZ4oEsyToWVHHMxvzsKE6qGs2ICMaaEywII9fwlOAyLjDSafmxNVGuHSsnCVvk3BR4KrSWJ6Z4v9dWfiBz3R3wZiV-8CftyyAdLThPAviQOrNeKQ0EGFIK3hwvEJxx-qKthtJzC-cj~p6Aq439Z4HjB05wVNy6OB3ZwvZloCujxWkrxFXPOCTZEKvidOfH3cSm-11JmoS7mvQe4vcAxvCbvLbqsicujr~njfaj93xQ8u0ahbs1aWNWmQyLPlFg__"
# 解压缩
tar -zxvf spaceranger-3.1.3.tar.gz
# 激活软件
source data/prostate/spaceranger-3.1.3/sourceme.bash
|
运行Space Ranger
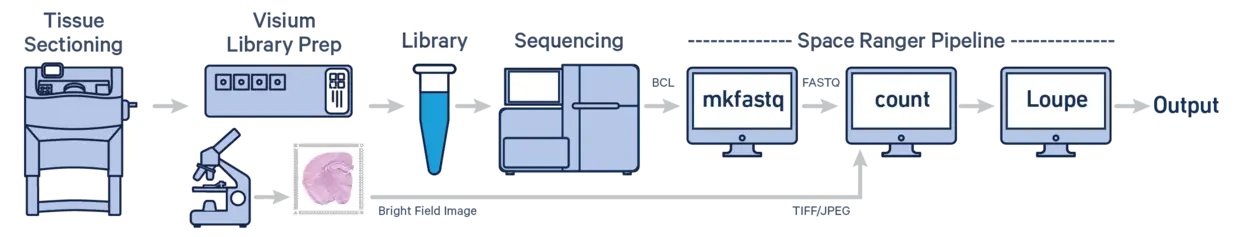
--id:指定输出的文件夹名称。
--transcriptome:指定参考基因组文件的路径。这里为刚才下载的人类参考基因组(GRCh38)
--probe-set:指定针对FFPE样本的捕获探针的文件路径
--fastqs:指定样本的测序文件的目录
--sample:指定 FASTQ 文件前缀,匹配文件名中的 SampleName 部分。【注意!】需与 FASTQ 文件名中的样本标识一致。
--image:指定样本的HE染色突变,可以是tiff或jpg格式
--slide:指定玻片编号(Visium 玻片的唯一 ID,如 V11J26-002)。
--area:选择玻片上的捕获区域(如 A1、B1 等)。
--localcores:指定使用的 CPU 核心数(本例为 16 个)。
--localmem:指定可用内存(GB)(本例为 48 GB)。
--create-bam:生成排序后的 BAM 文件(包含比对结果)。用于下游自定义分析(如变异检测或深度过滤)。
运行该命令要耗费数小时的时间!
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
|
wd="/mnt/d/10xVisium"
cd ${wd}
# 【关键步骤!】
spaceranger count --id=PC1\
--transcriptome=data/prostate/refdata-gex-GRCh38-2020-A\
--probe-set=data/prostate/Visium_FFPE_Human_Prostate_Acinar_Cell_Carcinoma_probe_set.csv\
--fastqs=data/prostate/Visium_FFPE_Human_Prostate_Acinar_Cell_Carcinoma_fastqs\
--sample=Visium_FFPE_Human_Prostate_Acinar_Cell_Carcinoma\
--image=data/prostate/Visium_FFPE_Human_Prostate_Acinar_Cell_Carcinoma_image.tif\
--slide=V11J26-002\
--area=B1\
--localcores=16\
--localmem=48\
--create-bam=true
|
Space Ranger的输出结果
输出的文件目录
Space Ranger的输出结果默认保存在{id_name}/outs文件夹下,其文件夹结构如下:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
|
(scanpy) D:\10xVisium>tree PC1/outs
卷 新加卷 的文件夹 PATH 列表
卷序列号为 0A60-29DF
D:\10XVISIUM\PC1\OUTS
├─analysis
│ ├─clustering
│ │ ├─gene_expression_graphclust
│ │ ├─gene_expression_kmeans_10_clusters
│ │ ├─gene_expression_kmeans_2_clusters
│ │ ├─gene_expression_kmeans_3_clusters
│ │ ├─gene_expression_kmeans_4_clusters
│ │ ├─gene_expression_kmeans_5_clusters
│ │ ├─gene_expression_kmeans_6_clusters
│ │ ├─gene_expression_kmeans_7_clusters
│ │ ├─gene_expression_kmeans_8_clusters
│ │ └─gene_expression_kmeans_9_clusters
│ ├─diffexp
│ │ ├─gene_expression_graphclust
│ │ ├─gene_expression_kmeans_10_clusters
│ │ ├─gene_expression_kmeans_2_clusters
│ │ ├─gene_expression_kmeans_3_clusters
│ │ ├─gene_expression_kmeans_4_clusters
│ │ ├─gene_expression_kmeans_5_clusters
│ │ ├─gene_expression_kmeans_6_clusters
│ │ ├─gene_expression_kmeans_7_clusters
│ │ ├─gene_expression_kmeans_8_clusters
│ │ └─gene_expression_kmeans_9_clusters
│ ├─pca
│ │ └─gene_expression_10_components
│ ├─tsne
│ │ └─gene_expression_2_components
│ └─umap
│ └─gene_expression_2_components
├─deconvolution
│ ├─deconvolution_k10
│ ├─deconvolution_k2
│ ├─deconvolution_k3
│ ├─deconvolution_k4
│ ├─deconvolution_k5
│ ├─deconvolution_k6
│ ├─deconvolution_k7
│ ├─deconvolution_k8
│ └─deconvolution_k9
├─filtered_feature_bc_matrix
├─raw_feature_bc_matrix
└─spatial
|
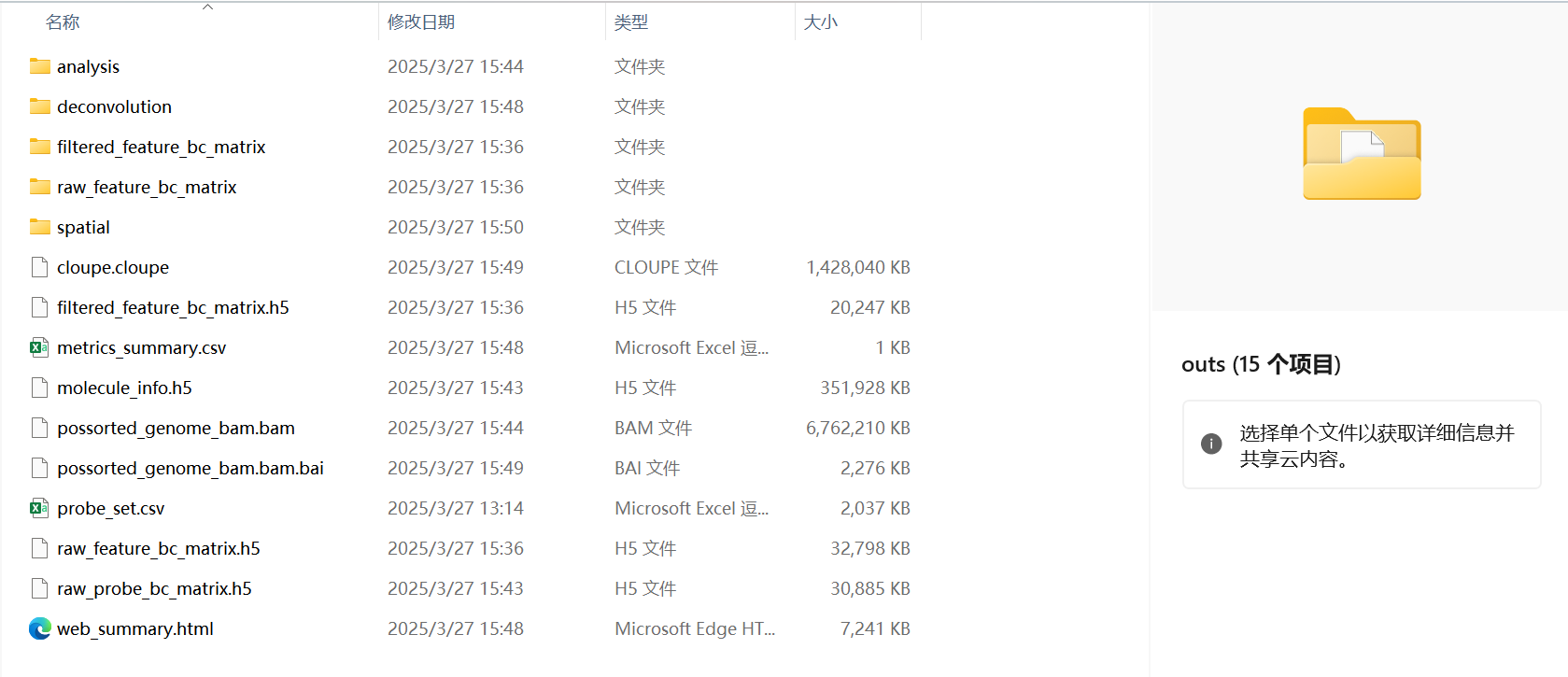
一些重要的文件
其中比较重要的文件包括:
analysis文件夹:存储了以默认参数进行的一些分析结果,包括K-means聚类,差异基因表达,以及降维的结果
filtered_feature_bc_matrix文件夹:标准的10x测序文件,可以用R来读取
filtered_feature_bc_matrix.h5:该文件完全包含了filtered_feature_bc_matrix文件夹里的所有信息,并且占用空间甚至更小。该格式适合使用scanpy进行读取
possorted_genome_bam.bam:文件是比对文件,后续可做RNA速率分析
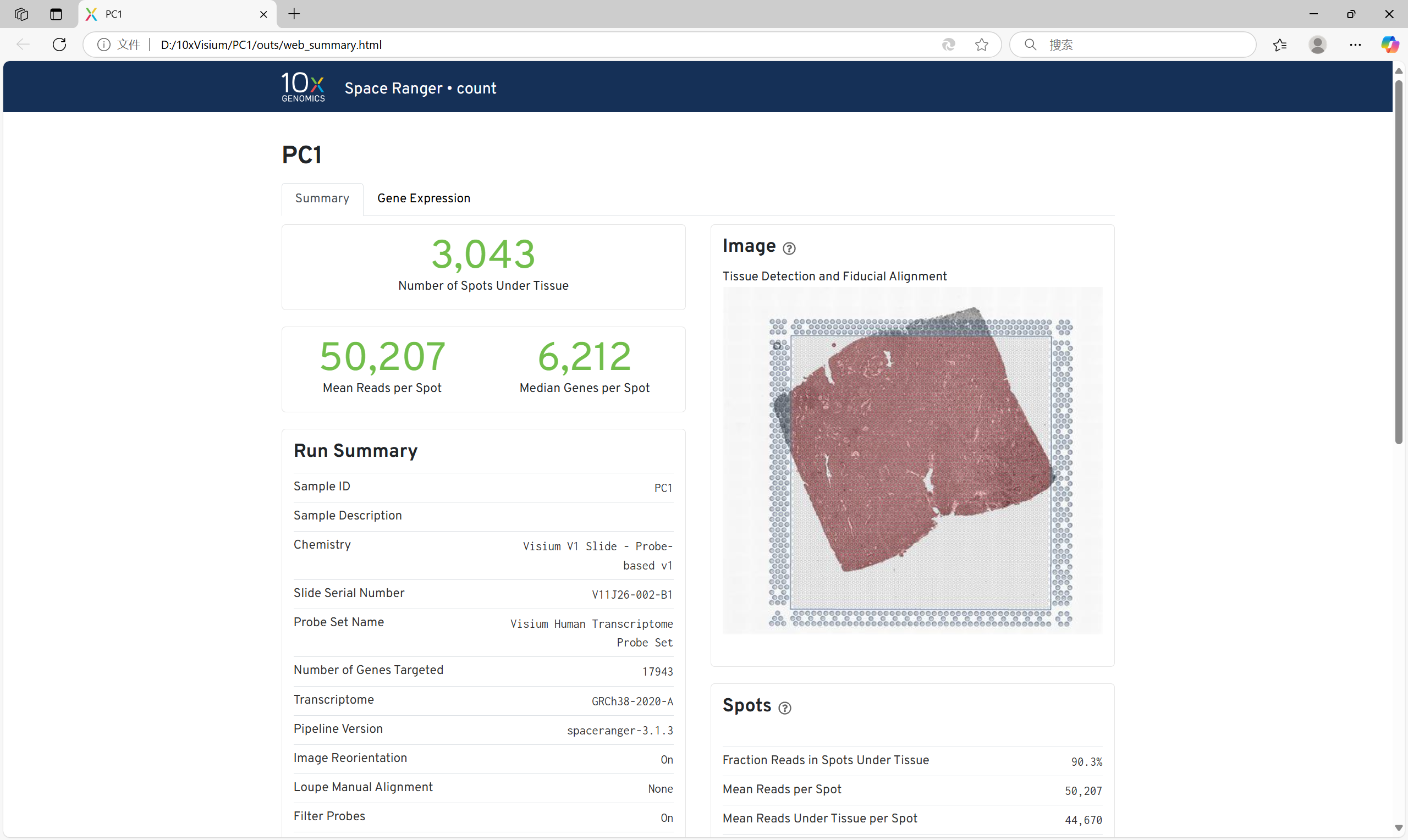
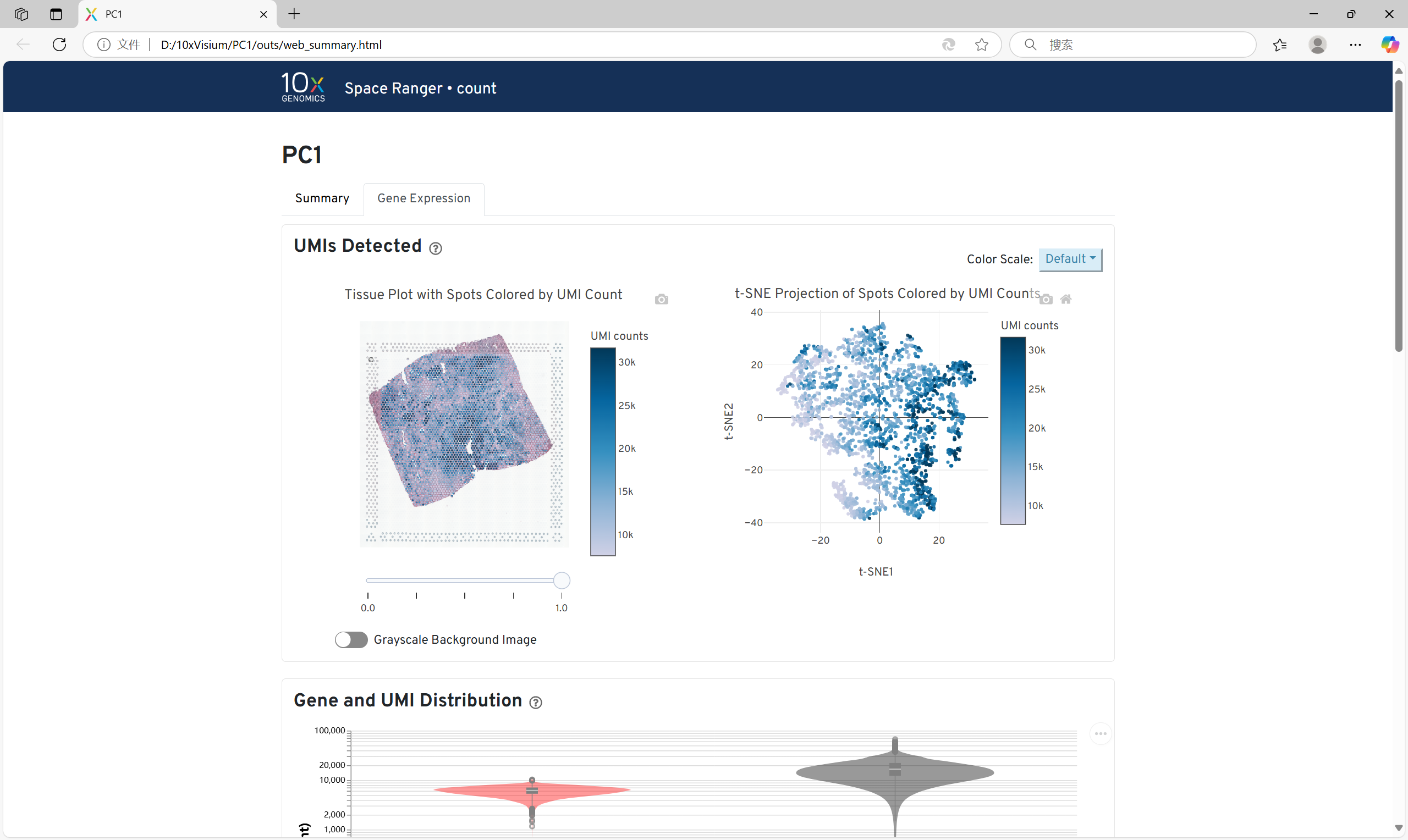
web_summary.html:一个网页版报告,可以查看空间转录的基本信息:
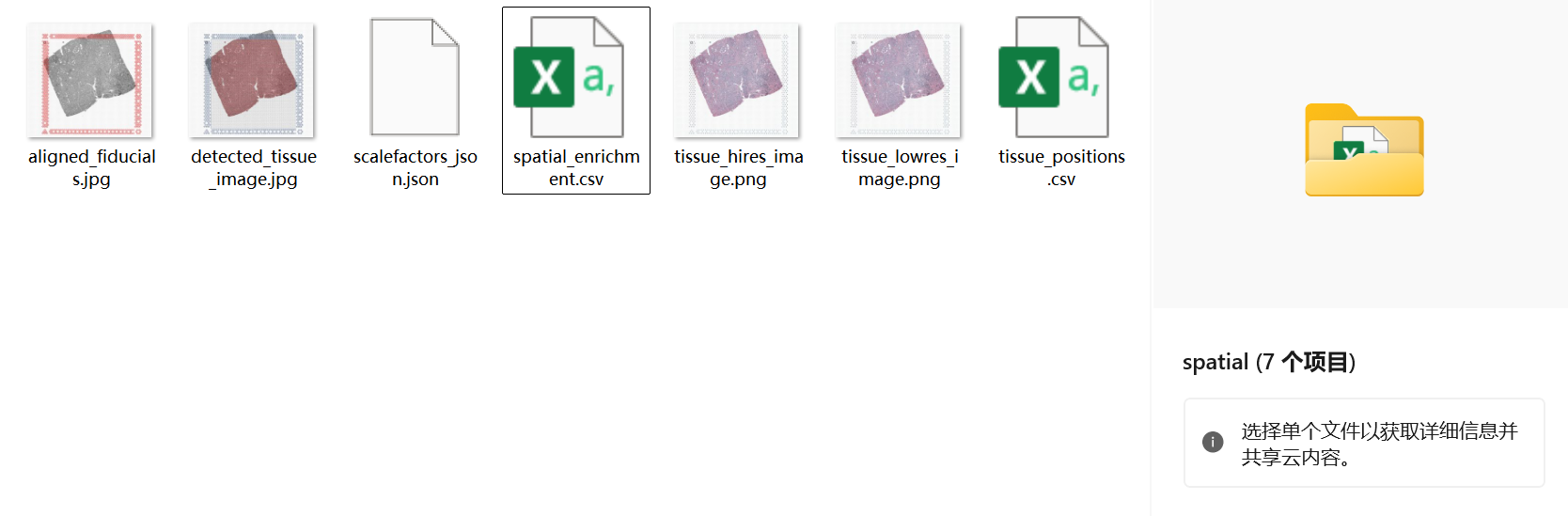
spatial文件夹:里面包含了组织的空间信息:
aligned_fiducials.jpg:验证基准点有无对齐,图像是否发生错位detected_tissue_image:高亮识别到的组织tissue_hires_image.png:高分辨率图像(high resolution)tissue_lowres_image.png:低分辨率图像(low resolution)scalefactors_json.json:记录了原始tiff图像转为两个png图像的缩放倍率tissue_positions:记录了每个spot的barcode、坐标以及在原始图片上的位置(像素x像素)spatial_enrichment.csv:记录了每个基因的空间分布情况,如空间自相关指数(Moran’s I 指数)、UMI计数、覆盖的spots数等等。