样本质量控制与过滤
在https://github.com/baidefeng/EasyMetagenome/tree/master/seq上下载四个原始文件:C1_1.fq.gz、C1_2.fq.gz、C2_1.fq.gz、C2_2.fq.gz,以及在https://github.com/baidefeng/EasyMetagenome/blob/master/result/metadata.txt上下载对应的metadata.txt。
| SampleID |
Group |
Replicate |
Sex |
Individual |
GSA |
CRR |
| C1 |
Cancer |
1 |
Male |
p136 |
CRA002355 |
CRR117732 |
| C2 |
Cancer |
2 |
Male |
p143 |
CRA002355 |
CRR117732 |
样本的测序结果
测序的fastq文件内容如下,分别代表正向测序和反向测序,上下每一个条目都是一一对应的。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
|
# C1_1.fq.gz
@SRR3586062.883556
CTTGGGGCTGCTGAGCTTCATGCTCCCCTCCTGCCTCAAGGACAATAAGGAGATCTTCGACAAGCCTGCAGCAGCTCGCATCGACGCCCTCATCGCTGAGG
+
CCCFFFFFHHHHHIJJJJJJJIJIJJJJGIJDGIJEIIJIJJJJJJJJIJJJJIJJIJJJJJHHHFFFFECEEEDDDDD?BDDDDDDBDDDDDDDDBBBDD
@SRR3586062.3376311
GACGGTGTCCTCAGGACCCTTCAGTGCCTTCATGATCTGCTCAGAGGTGATGGAGTCACGGACGAGATTCGTCGTGTCAGCACGTAGGATGCGGTCGCCTG
+
@@@DDDDAFF?DF;EH+ACHIIICHDEHGIGBFE@GCGDGG?D?G@BGHG@FHCGC;CC:;8ABH>BECCBCB>;8ABCCC@A@#################
# C1_2.fq.gz
@SRR3586062.883556
AAGCCAGGACCCGAGTAGTCTCGGCCAGCATAGTACTCCATACGCCAGCCCTTGGGCGCAGCTTCGAGAAGGGCTTTCTCCTCAGCGATGAGGGCGTCGAT
+
CCCFFFFFHHHHHJJHIJEHIJJHIIIIIIJJJHIJGIJIGIJJJJGGCHFEGCHGHGFBCDCDDBD?ADDDDDDDDDDDDCDDDDD@BDCCCBDD<BD><
@SRR3586062.3376311
TGGCGTGCAGTTCAATACCCTCCTCGACACCGTCGTCGTCGTGCGTGTCATCGAGGGTGGCCCCAGCGAGCGTGCAGGCCTGCAGCCAGGCGACCGCATCT
+
=8+=?=)0<+CDDEAEEA<C>FD>F<EEDADDBD)@DA7BADDD8CDA(7.;@A@@>=;:?==??88;>9>>;9-<>>>>A82<AA###############
|
软件下载
下载质量评估所需的软件:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
|
wd=PATH/TO/WORKINGDIR
cd $wd
#### 这里选择在kneaddata环境中安装质量控制所需软件
conda activate kneaddata
# fastqc 0.12.1 kneaddata环境自带,无需下载
# fastp 0.23.2
conda install fastp
# multiqc 1.27
conda install multiqc
# rush 0.6.0
conda install rush
# 将python升级到3.9及以上版本
conda install python=3.9
|
测序结果质量评估与筛选
使用fastqc或multiqc进行质量评估(可选)
1
2
3
4
5
|
# 使用fastqc进行质量评估,-t 1代表设置线程数为1
fastqc seq/*.gz -t 1
# 使用multiqc进行多样本汇总,指定测序样本文件夹和结果输出文件夹
multiqc -d seq/ -o result/qc
|
使用fastp进行质量评估与筛选
可以使用fastp进行数据质量评估与筛选,并且用rush工具进行并行管理。该命令含义为:使用tail命令读取metadata.txt文件,-n +02指的是从第二行读到文件末尾。将结果传递给cut -f1命令,意为读取每行的第一个字段,这里即为C1和C2。将结果传给rush -j 2 "command",表示同时处理2个样本。双引号内为fastp命令:-i和-I分别对应Read1(前向测序结果)和Read2(反向测序结果),-j和-h分别指定生成的json文件和html文件。> temp/qc/{}.log 2>&1指将标准输出流1重新定向至temp/qc/{}.log文件,标准错误输出2重定向至标准输出1,即上文所述的temp/qc/{}.log文件。此外,{}里面就是指rush传进来的参数。
fastp的参数可调,在不指定参数的情况下将以默认参数进行过滤:
-q 15:碱基质量的阈值为15,<15的碱基为不合格碱基。
-u 40:不合格碱基占比阈值为40%,不合格碱基>40%将被过滤。
-l 15:长度<15bp的reads将被过滤
-y 30:reads复杂度<30时将被过滤。reads的复杂度指一个碱基与下一个相邻碱基不同的碱基个数占比。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
|
tail -n +2 metadata.txt | cut -f1 | rush -j 2 \
"fastp -i seq/{}_1.fq.gz -I seq/{}_2.fq.gz \
-j temp/qc/{}_fastp.json -h temp/qc/{}_fastp.html \
-o temp/qc/{}_1.fastq -O temp/qc/{}_2.fastq \
> temp/qc/{}.log 2>&1 "
# 提取temp/qc/${i}.log里的关键信息,生成质控的摘要,即每个fastq文件的原始数据条目数量以及滤过后的数据条目数量
echo -e "SampleID\tRaw\tClean" > temp/fastp
for i in `tail -n+2 metadata.txt | cut -f1`; do
echo -e -n "$i\t" >> temp/fastp
grep 'total reads' temp/qc/${i}.log | uniq | cut -f2 -d ':' | tr '\n' '\t' >> temp/fastp
echo "" >> temp/fastp
done
sed -i 's/ //g;s/\t$//' temp/fastp
|
使用kneaddata过滤宿主基因
使用kneadata将测序数据中的宿主基因给过滤掉。这一步需要使用到宿主的参考基因组,以及基于参考基因组生成的索引文件。对于每对fastq文件都会生成相应的四个文件:{}.paired_1.fastq、{}.paired_2.fastq、{}.unmatched_1.fastq、{}.unmatched_2.fastq,其中前两个文件记录了过滤后还能配对的记录,后两个文件记录了过滤后虽然幸存,但是无法配对的记录。最后生成的result/qc/sum.txt以表格的形式展示如下:
| Sample |
raw pair1 |
decontaminated human pair1 |
decontaminated human pair2 |
| C1 |
71279.0 |
71076.0 |
71076.0 |
| C2 |
54008.0 |
45882.0 |
45882.0 |
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
|
# 创建并进入kneaddata环境
conda create -c bioconda -n kneaddata kneaddata
conda activate kneaddata
# 从NCBI上下载人类参考基因组CRCh38.(3.11Gb)
wget -c https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/405/GCF_000001405.40_GRCh38.p14/GCF_000001405.40_GRCh38.p14_genomic.fna.gz
# 使用bowtie2生成索引文件,这个步骤需要消耗数小时的时间。对于人类基因组可以直接下载生成好的索引文件,而对于其它小众物种的基因组,则仍需要自行使用bowtie2生成。
bowtie2-build GCF_000001405.40_GRCh38.p14_genomic.fna /mnt/d/metagenome/databases/kneaddata/huma
n --threads 10
# 指定数据库的存放位置
db=/mnt/d/metagenome/databases
# 使用kneaddata去宿主。这里需要对原始测序文件进行处理:同一个样本需要有相同的前缀名称,在名称最后加上“/1”或“/2”分别表示是前向测序还是反向测序。这里通过sed命令将所有{}_1.fastq的每条记录的名称末尾加上“/1”,同时将所有{}_2.fastq的每条记录的名称末尾加上“/2”,之后将处理好的fastq文件传给kneaddata进行过滤。
tail -n+2 metadata.txt | cut -f1 | rush -j 2 \
"sed '1~4 s/^@\(.*\)$/@\1\/1/' temp/qc/{}_1.fastq > /tmp/{}_1.fastq; \
sed '1~4 s/^@\(.*\)$/@\1\/2/' temp/qc/{}_2.fastq > /tmp/{}_2.fastq; \
kneaddata -i1 /tmp/{}_1.fastq -i2 /tmp/{}_2.fastq \
-o temp/hr --output-prefix {} \
--bypass-trim --bypass-trf --reorder \
--bowtie2-options '--very-sensitive --dovetail' \
-db ${db}/kneaddata/human\
--remove-intermediate-output -v -t 4; \
rm /tmp/{}_1.fastq /tmp/{}_2.fastq"
# 结果改名
rename 's/paired_//' temp/hr/*.fastq
# 质控结果汇总,即读取“{}.log”文件
kneaddata_read_count_table --input temp/hr \
--output temp/kneaddata.txt
# 筛选重点结果列,我们只关心1、2、5、6列的信息
cut -f 1,2,5,6 temp/kneaddata.txt | sed 's/_1_kneaddata//' > result/qc/sum.txt
# 查看内容
cat result/qc/sum.txt
|
物种组成和功能分析
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
|
# 安装biobakery/humann3环境
conda create --name biobakery3 python=3.7
conda activate biobakery3
conda config --add channels defaults
conda config --add channels bioconda
conda config --add channels conda-forge
conda config --add channels biobakery
conda install humann -c biobakery
db=/mnt/d/metagenome/databases
# 下载三个数据库。下载完毕后会在配置文件中自动修改数据库的存放位置。
# upgrade your pangenome database: 微生物物种核心基因(约15Gb)
humann_databases --download chocophlan full ${db}/humann --update-config yes
# upgrade your protein database: 功能基因diamond索引(约34Gb)
humann_databases --download uniref uniref90_diamond ${db}/humann --update-config yes
# upgrade your annotations database: 输助比对数据库(约3Gb)
humann_databases --download utility_mapping full ${db}/humann --update-config yes
# 在用humann3分析之前,需要将双端测序的两个文件合并为一个文件
for i in `tail -n+2 metadata.txt | cut -f1`; do
cat temp/hr/${i}_?.fastq \
> temp/concat/${i}.fq; done
# 在MetaPhlAn4 官方网站下载最新的数据库文件,解压到数据库。然后修改metaphlan_database.cfg文件,将bowtie2db和mpa_pkl参数的值改为数据库文件的路径。如果不写--offline参数,则命令运行时会自动下载。(约20Gb)
# 该命令同时运行了metaphlan和humann命令,生成的关键文件包括物种相对丰度文件、通路丰度文件以及基因家族丰度文件。
tail -n+2 metadata.txt | cut -f1 | rush -j 2 \
"humann --input temp/concat/{}.fq\
--output temp/humann3/ \
--threads 8\
--metaphlan-options '-t rel_ab --sample_id {} --offline'"
# 将${}_metaphlan_bugs_list.tsv文件移动位置
for i in $(tail -n+2 metadata.txt | cut -f1); do
mv temp/humann3/${}_humann_temp/${}_metaphlan_bugs_list.tsv temp/humann3/
done
# 合并表格。注意,合并后tsv文件的第二行恒定为“#metaphlan4”,以便接下来的metaphlan_to_stamp.pl能够正确识别。
merge_metaphlan_tables.py temp/humann3/*_metaphlan_bugs_list.tsv | \
sed 's/_metaphlan_bugs_list//g' |\
tail -n+2 | sed '1 s/clade_name/ID/' |\
sed '2i #metaphlan4'> result/metaphlan4/taxonomy.tsv
# metaphlan4较2增加更多unclassified和重复结果,用sort和uniq去除:在https://github.com/LangilleLab/microbiome_helper/blob/master/metaphlan_to_stamp.pl上下载metaphlan_to_stamp.pl文件,放到~/miniconda3/envs/biobakery3/bin文件夹内。
chmod +777 ~/miniconda3/envs/biobakery3/bin/metaphlan_to_stamp.pl
metaphlan_to_stamp.pl result/metaphlan4/taxonomy.tsv \
|sort -r |\
uniq > result/metaphlan4/taxonomy.spf
# STAMP不支持unclassified,需要过滤掉再使用。使用该文件可进行stamp分析
grep -v 'unclassified' result/metaphlan4/taxonomy.spf > result/metaphlan4/taxonomy2.spf
# 将文件的最后一行移动至第一行,以便STAMP软件可以正确识别。
(tail -n 1 "result/metaphlan4/taxonomy2.spf" && head -n -1 "result/metaphlan4/taxonomy2.spf") \
> result/metaphlan4/taxonomy3.spf
|
使用STAMP进行绘图
1、STAMP软件的安装
在STAMP官网STAMP - Bioinformatics Software下载安装包,按照提示安装即可。
2、使用STAMP软件打开*.spf文件
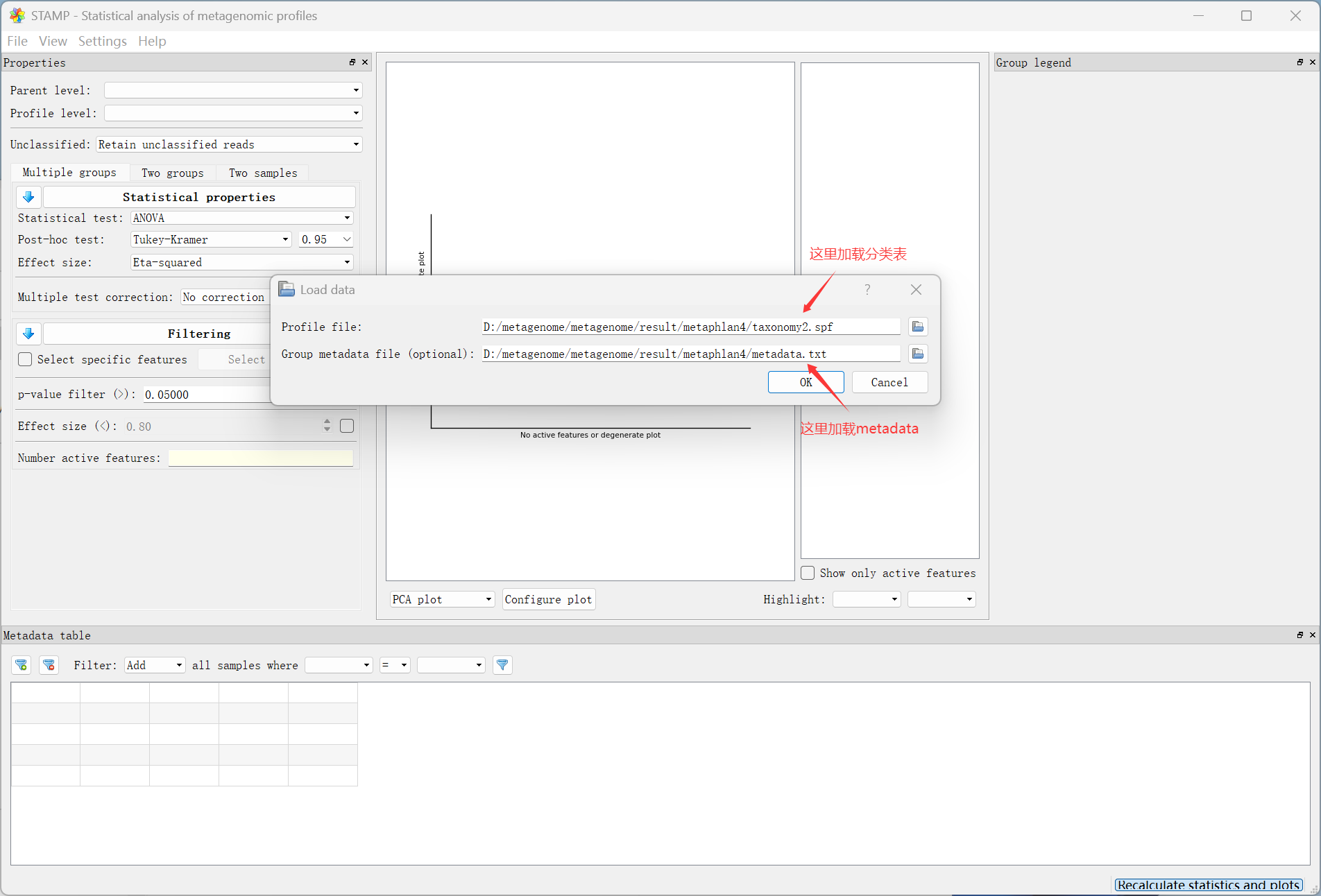
打开软件,点击File、Load data,然后分别选择上文中的taxonomy3.spf以及metadata.txt即可进行绘图。不过我们的简化数据只有两个样本,在这里我们下载教程中所给的完整的具有12个样本的taxonomy.spf和metadata.txt作为示例文件。
1
2
|
# https://github.com/baidefeng/EasyMetagenome/blob/master/result12/metaphlan4/taxonomy.spf
# https://github.com/baidefeng/EasyMetagenome/blob/master/result12/metadata.txt
|
1
2
|
# 过滤掉Unclassified结果
grep -v 'unclassified' result/metaphlan4/taxonomy.spf > result/metaphlan4/taxonomy2.spf
|
点击File、Load data,然后分别选择上文中的taxonomy2.spf以及metadata.txt。
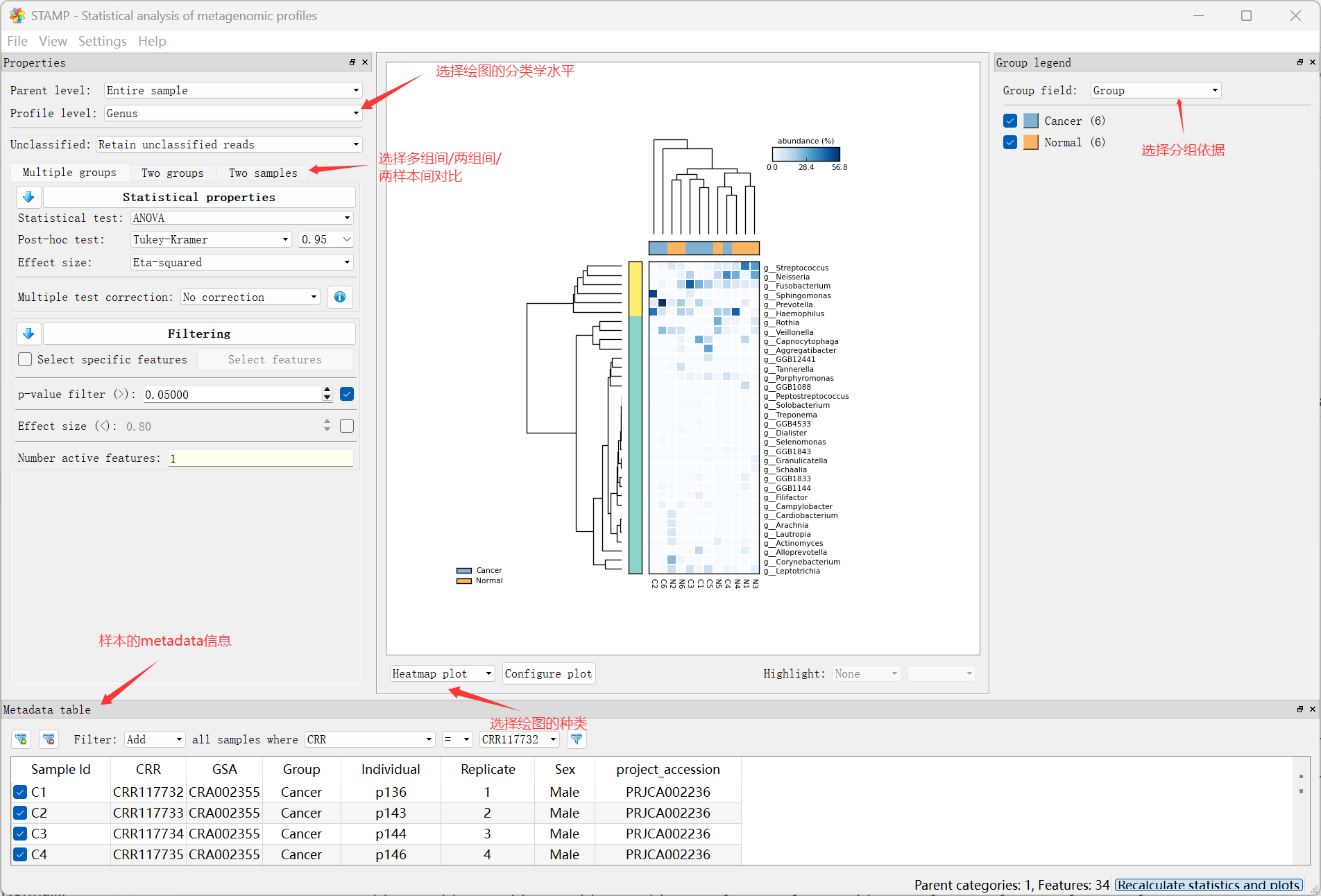
点击File、Save plot即可导出。
使用humann3获得样本的功能数据
这步在上述代码中已经完成,每个样本的功能数据已经记录在{}_pathabundance.tsv中。
合并样品功能数据表
1
2
3
4
5
6
|
humann_join_tables --input temp/humann3 \
--file_name pathabundance \
--output result/humann3/pathabundance.tsv
# 将样本名后面的后缀去掉,更加美观
sed -i 's/_Abundance//g' result/humann3/pathabundance.tsv
|
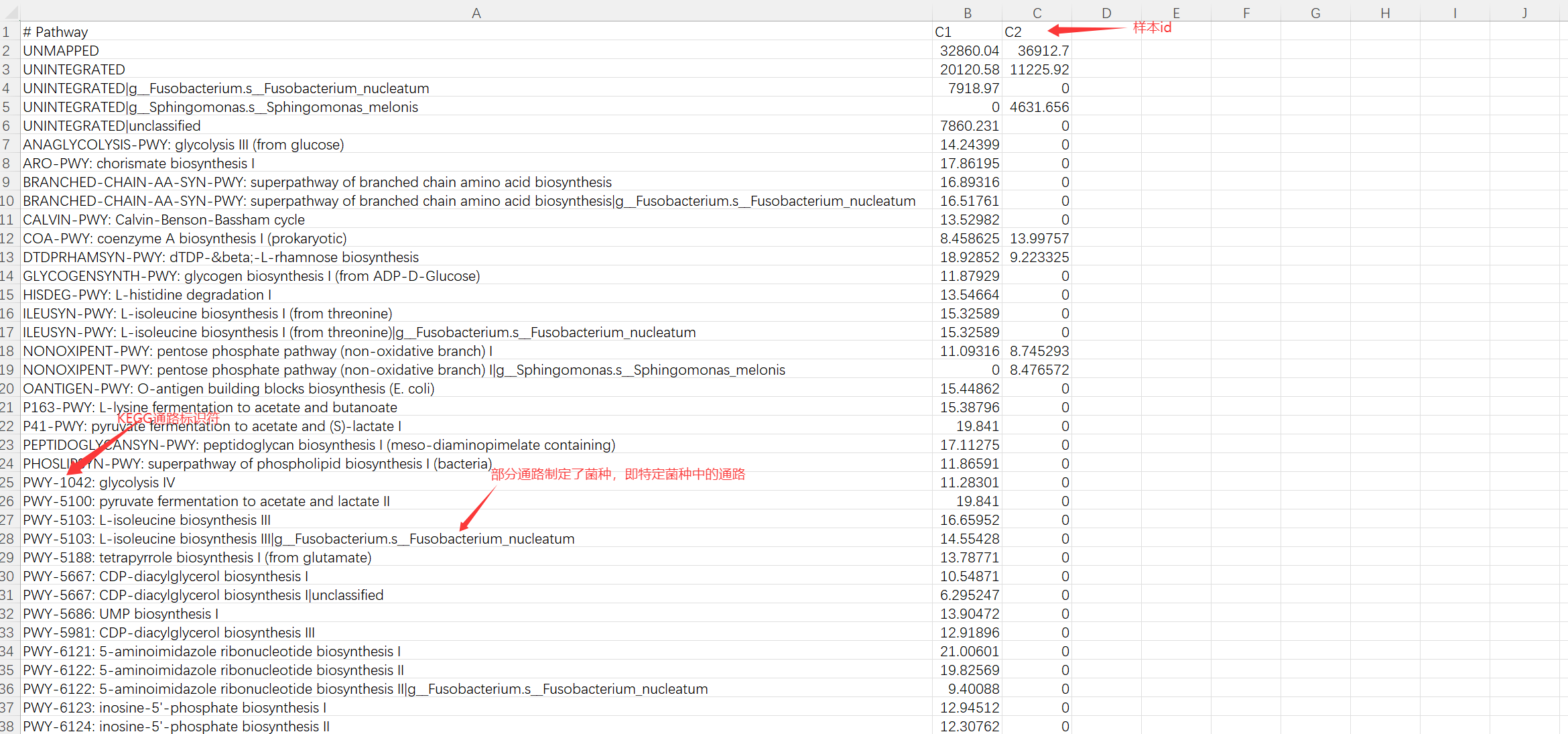
合并后的文件可用excel打开:
转化为相对丰度relab或百万比cpm
1
2
3
4
|
humann_renorm_table \
--input result/humann3/pathabundance.tsv \
--units relab \
--output result/humann3/pathabundance_relab.tsv
|
拆分表格
将丰度表拆分成功能和对应物种表(stratified)和功能组成表(unstratified)。该步骤采用humann_split_stratified_table命令,其作用是将分层功能丰度表拆分成两个独立的表格:
如下图所示,左表为分层功能丰度表,右上即为unstratified表,记录了功能的总体丰度;右下是stratified表,仍然保留了该通路每种物种的贡献。
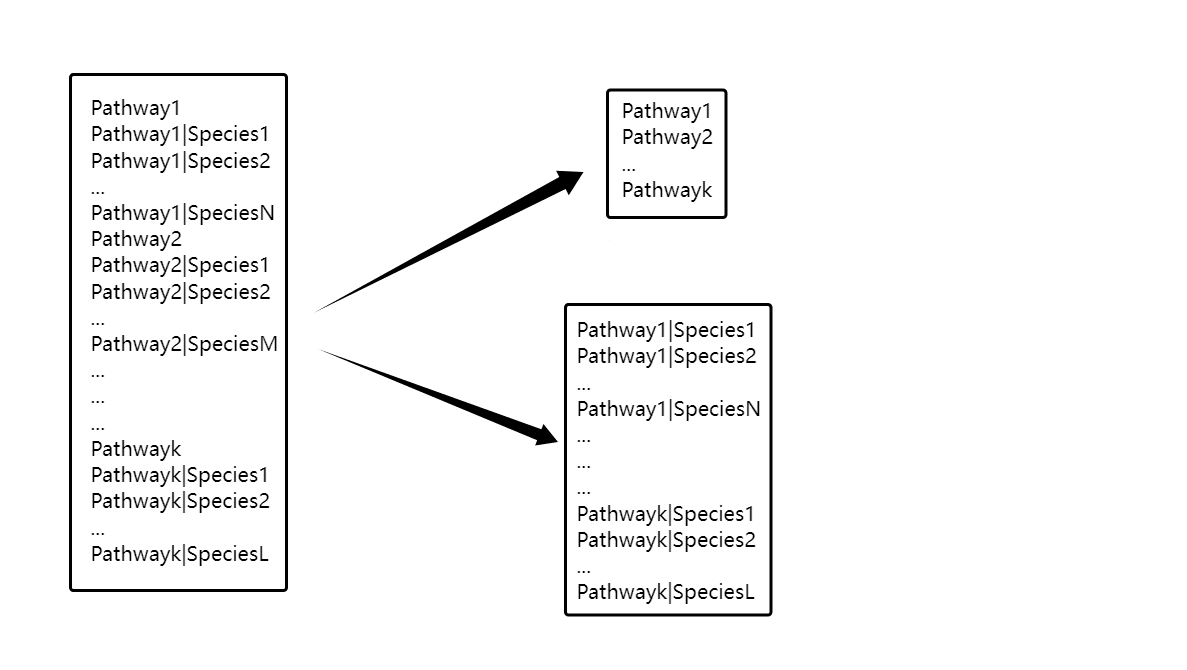
以下命令会将pathabundance_relab.tsv拆分成pathabundance_relab_stratified.tsv和pathabundance_relab_unstratified.tsv两个文件
1
2
3
|
humann_split_stratified_table \
--input result/humann3/pathabundance_relab.tsv \
--output result/humann3/
|
构建pcl文件,进行差异分析与条形图绘制
接下来,从pathabundance_relab_unstratified.tsv文件出发,给其第二行加上metadata信息,最终生成pathabundance_relab_unstratified.pcl
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
|
#### 【关键步骤】给上一步生成的stratified表的第二行加上metadata信息,使之变成pcl文件(*.pcl是接下来分析和画图的关键文件!)
# 提取样品列表
head -n1 result/humann3/pathabundance_relab_stratified.tsv | sed 's/# Pathway/SampleID/' | tr '\t' '\n' > temp/header
# 对应分组,本示例分组为第3列($3),根据实际情况修改。
awk 'BEGIN{FS=OFS="\t"}NR==FNR{a[$1]=$3}NR>FNR{print a[$1]}' metadata.txt temp/header | tr '\n' '\t'|sed 's/\t$/\n/' > temp/group
# 合成样本、分组+数据
cat <(head -n1 result/humann3/pathabundance_relab_stratified.tsv) temp/group <(tail -n+2 result/humann3/pathabundance_relab_stratified.tsv) \
> result/humann3/pathabundance_relab_stratified.pcl
#### 对上一步生成的pcl文件,进行表达差异分析与柱状图的绘制
# 使用humann_associate命令进行差异分析,其中需要指定metadata的名称和类型,以及p值。
pcl=result/humann3/pathabundance_relab_stratified.pcl
humann_associate --input ${pcl} \
--focal-metadatum Replicate --focal-type categorical \
--last-metadatum Replicate --fdr 0.05 \
--output result/humann3/associate.txt
wc -l result/humann3/associate.txt
# 指定感兴趣的通路名称,使用humann_barplot绘制分层条形图
path=PWY-6277
humann_barplot \
--input ${pcl} --focal-feature ${path} \
--focal-metadata Replicate --last-metadata Replicate \
--output result/humann3/barplot_${path}.pdf --sort sum metadata
|
这里演示数据的样本量过少,且都是肿瘤组的,无法分析出差异结果。故这里下载其他的演示数据进行演示。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
|
# 下载别人的*.pcl文件
wget -c http://www.imeta.science/github/EasyMetagenome/result/humann2/hmp_pathabund.pcl
cp hmp_pathabund.pcl result/humann3/hmp_pathabund.pcl
# 进行差异分析。这里指定使用Group进行分组
pcl=result/humann3/hmp_pathabund.pcl
humann_associate --input ${pcl} \
--focal-metadatum Group --focal-type categorical \
--last-metadatum Group --fdr 0.05 \
--output result/humann3/associate.txt
wc -l result/humann3/associate.txt
# 选择感兴趣的通路进行绘图
path=P163-PWY
humann_barplot \
--input ${pcl} --focal-feature ${path} \
--focal-metadata Group --last-metadata Group \
--output result/humann3/barplot_${path}.pdf --sort sum metadata
|
最终绘图结果如下。在本图中,可以看到与L-赖氨酸发酵有关的通路P163-PWY在Tongue_dorsum组中富集,而对该通路贡献最多的细菌主要是*Fusobacterium periodonticum*。
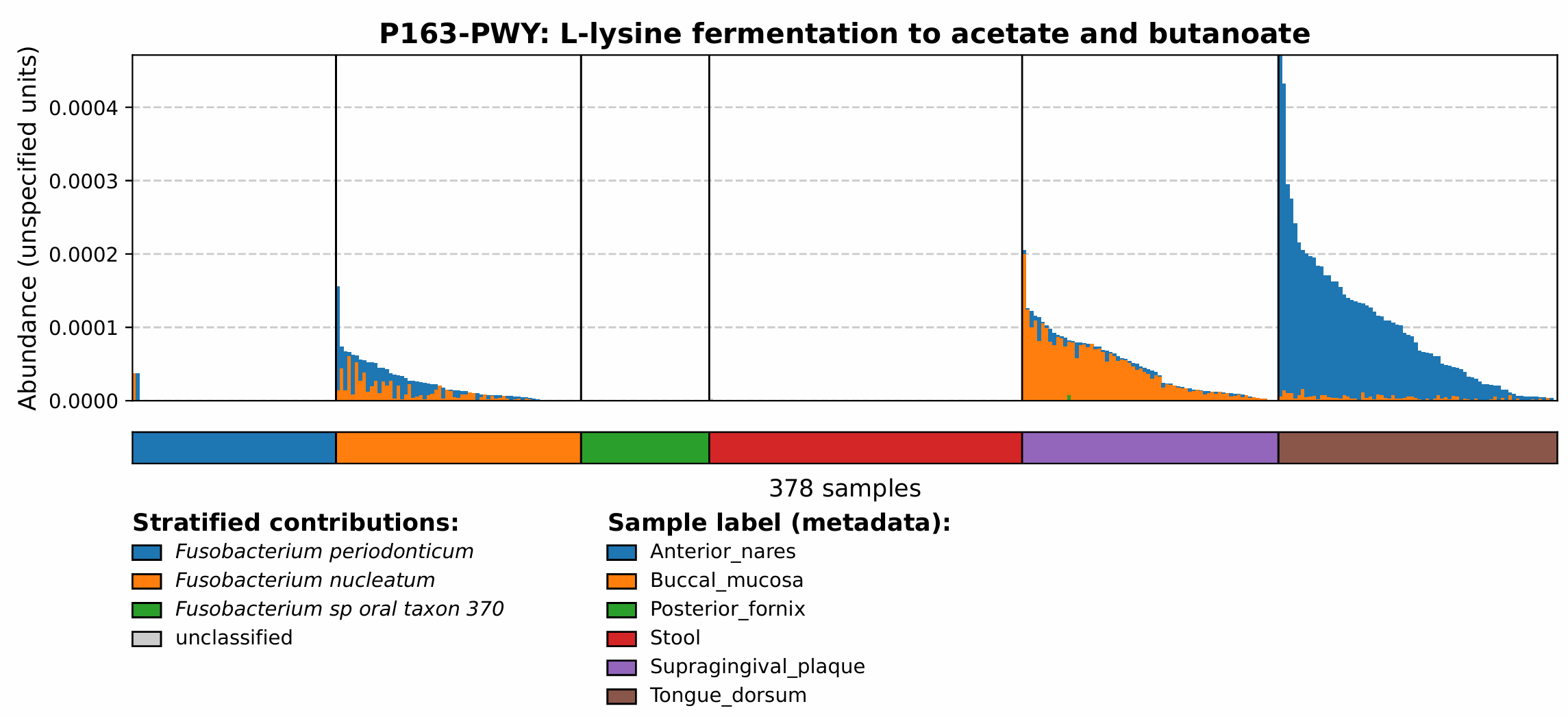
KEGG注释
如果不想用程序内置的通路映射文件,可以使用humann_regroup_table命令,指定感兴趣的注释系统,重新生成自己的pathabundance.tsv
HUMAnN3默认使用UniRef90数据库聚类基因家族(蛋白质序列),但这些家族本身没有直接的功能注释。而通过humann_regroup_table命令,可以通过预定义的映射文件(如 uniref90_ko),将每个 UniRef90 ID 的丰度合并到对应的功能条目中(如 KO 编号)。根据分析的需要,可以选择不同的注释系统:
- KO:适合代谢通路分析(如碳水化合物代谢)。
- EC:适合酶活性分析(如酶的催化功能)。
- eggNOG:提供更广泛的注释(如分子功能、生物过程)。
这里命令运行所需要的映射文件已经在前述的数据库下载中下载完毕了。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
|
# 转换基因家族为KO(uniref90_ko),可选eggNOG(uniref90_eggnog)或酶(uniref90_level4ec)
for i in `tail -n+2 metadata.txt|cut -f1`;do
humann_regroup_table \
-i temp/humann3/${i}_genefamilies.tsv \
-g uniref90_ko \
-o temp/humann3/${i}_ko.tsv
done
# 合并,并修正样本名
humann_join_tables \
--input temp/humann3/ \
--file_name ko \
--output result/humann3/ko.tsv
sed -i '1s/_Abundance-RPKs//g' result/humann3/ko.tsv
|
接下来可以对ko.tsv进行类似的操作,即拆分表格-构建pcl文件-差异分析与条形图绘制。
物种树的绘制
想要把样本中存在的微生物按照种属关系绘制成树形图,可以使用GraPhlAn软件。GraPhlAn是一个软件工具,可用于生成分类树和系统发育树的高质量圆形表示形式。 它着重于系统发育和分类学驱动的研究的简洁、整合、信息丰度信,输出结果图片可直接用于高水平文章发表使用。
通过export2graphlan.py的python程序,可以将MetaPhlAn4的输出结果转化成可以被GraphlAn读取的形式。运行该程序后会生成两个文件:
*.tree.txt:保留了特征表的第一列,即各分类学水平的物种清单。*.annot.txt:绘图一些参数,以及物种的丰度信息。需要注意的是,这里的丰度是所有样本的均值。
通过graphlan_annotate.py可以将两个txt文件融合成一个*.xml文件,此时graphlan.py即可通过该文件进行图形的绘制。
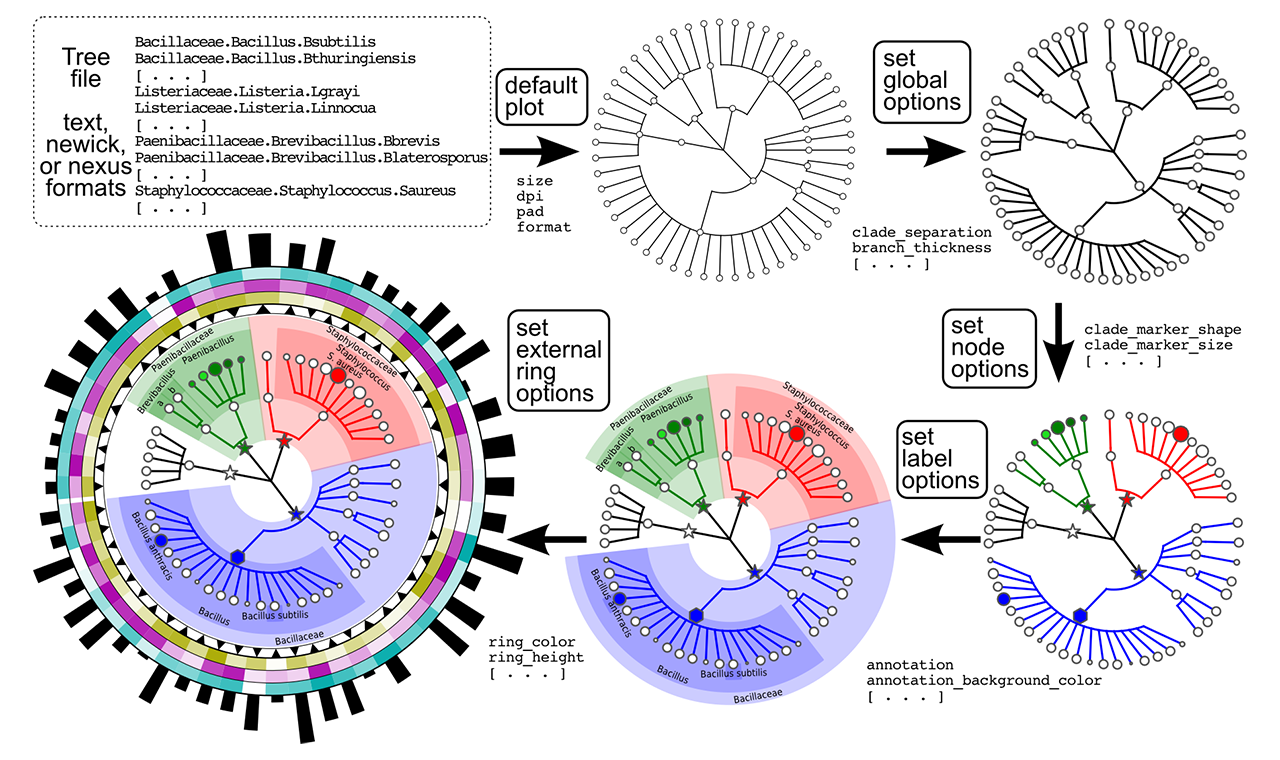
下载GraPhlAn程序包以及相关程序
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
|
# 创建GraPhlAn环境
conda create -n Graphlan
conda activate Graphlan
# 下载GraphlAn程序
conda install graphlan
# 下载格式转化程序,可以很方便的将LEfSe、MetaPhlAn4结果转化为GraPhlAn的输入文件
conda install export2graphlan
# 需要额外下载python库:biopython
conda install biopython
# 下载R语言核心以运行R脚本
conda install r-base
conda install r-optparse
|
绘图
绘图方式1
在运行export2graphlan.py的时候可以调节一些绘图的关键性参数:
--annotations:指定哪些分类层级(taxonomic levels)在树中被注释。参数值为层级编号,例如 3,4 表示对第3层和第4层的分类单元添加注释(如纲、目等)。需结合输入文件的分类层级结构来确定具体层级。--external_annotations:指定哪些分类层级使用外部图例注释。参数值为层级编号(如 7),表示第7层的分类单元将使用独立图例标注。这可以用于突出显示特定层级(如种或株)。--abundance_threshold:设置最低丰度阈值,低于此值的分类单元将不被注释。需结合输入文件的丰度计算方式确认。--most_abundant:指定保留的最丰富的分类单元数量(默认 10)。如果设置为一个很大的数,如1000,则代表给所有节点进行标注。--least_biomarkers:设置最小生物标志基因数阈值。确保每个分类节点至少包含 10 个生物标志基因(如 Metaphlan 的标记基因),避免低置信度的分类结果被保留。
更多绘图参数可以参见官方Github:https://github.com/segatalab/export2graphlan
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
|
# 下载示例文件
wget -c https://github.com/baidefeng/EasyMetagenome/raw/refs/heads/master/result12/metaphlan4/taxonomy.tsv
mv taxonomy.tsv result/metaphlan4/taxonomy.tsv
# 【关键步骤!】将taxonomy.tsv转化成*.tree.txt与*.annot.txt
export2graphlan.py --skip_rows 1,2 \
-i result/metaphlan4/taxonomy.tsv \
--tree temp/merged_abundance.tree.txt \
--annotation temp/merged_abundance.annot.txt \
--most_abundant 50 \
--abundance_threshold 10 \
--least_biomarkers 3 \
--annotations 1,2,3,4,5,6 \
--external_annotations 7
# 生成*.xml文件
graphlan_annotate.py --annot temp/merged_abundance.annot.txt \
temp/merged_abundance.tree.txt temp/merged_abundance.xml
# 输出pdf,这里会输出三个pdf文件,即主图,以及两个图注,后续可以使用Adobe Illustrator进行拼接
graphlan.py temp/merged_abundance.xml result/metaphlan4/graphlan.pdf \
--external_legends
|
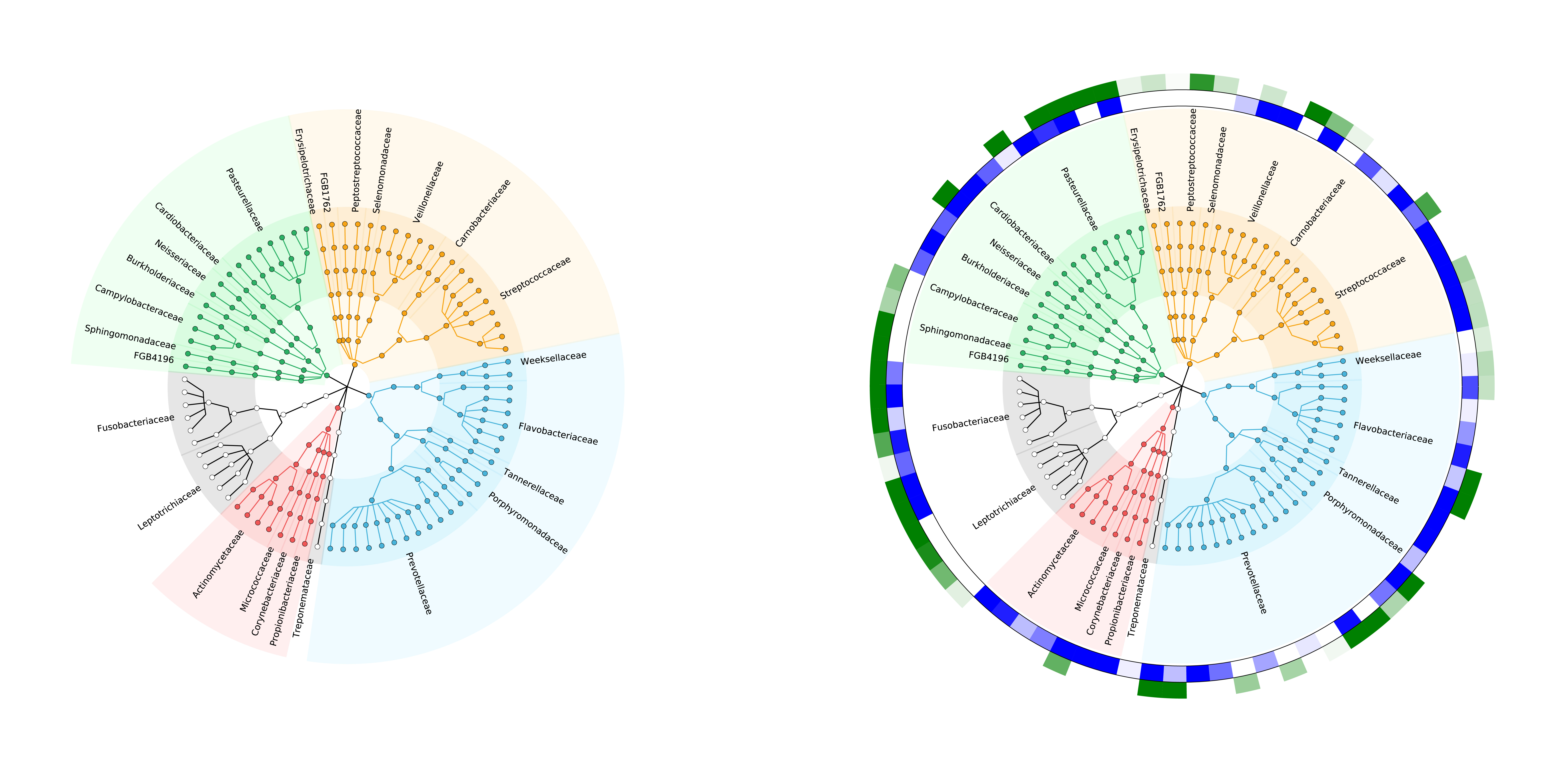
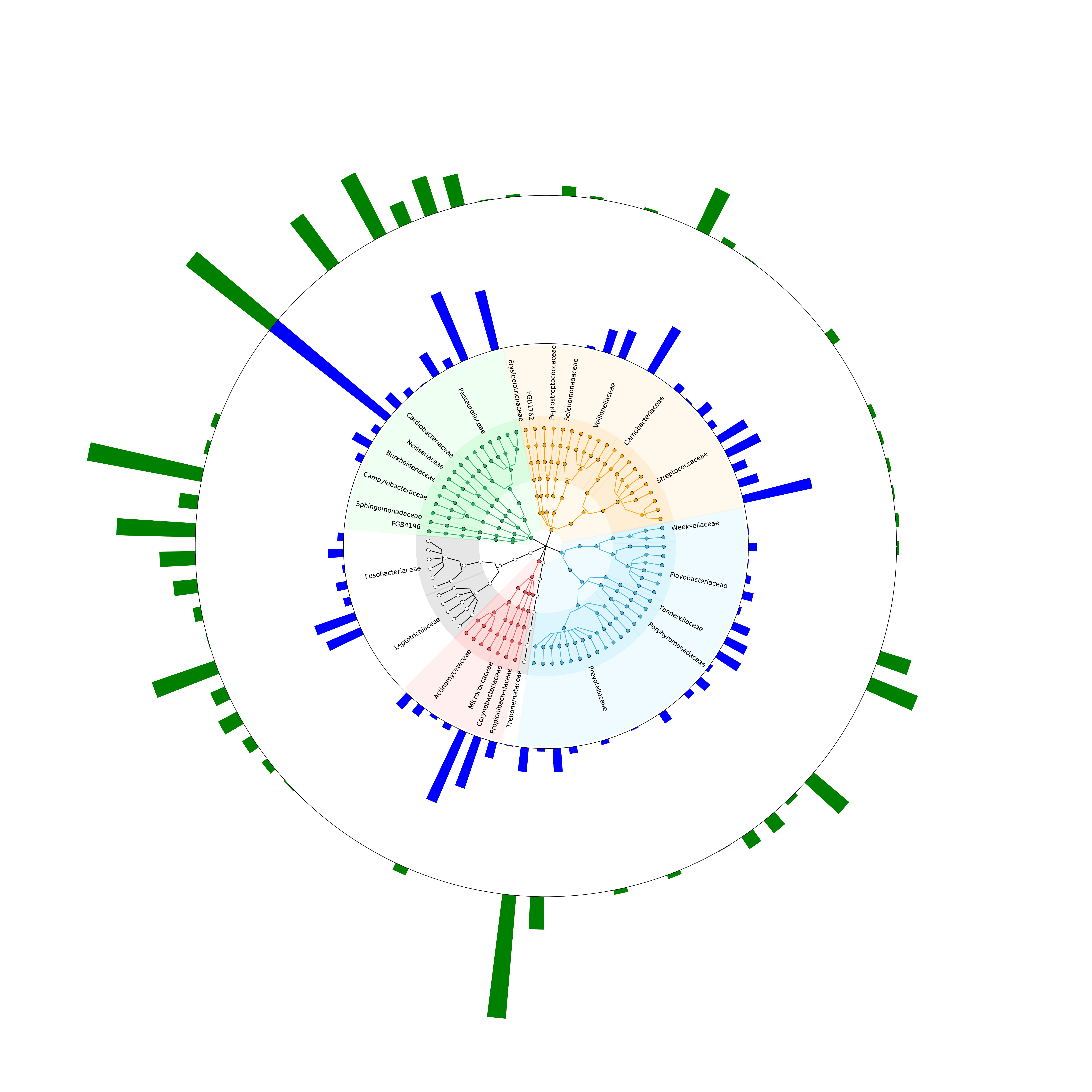
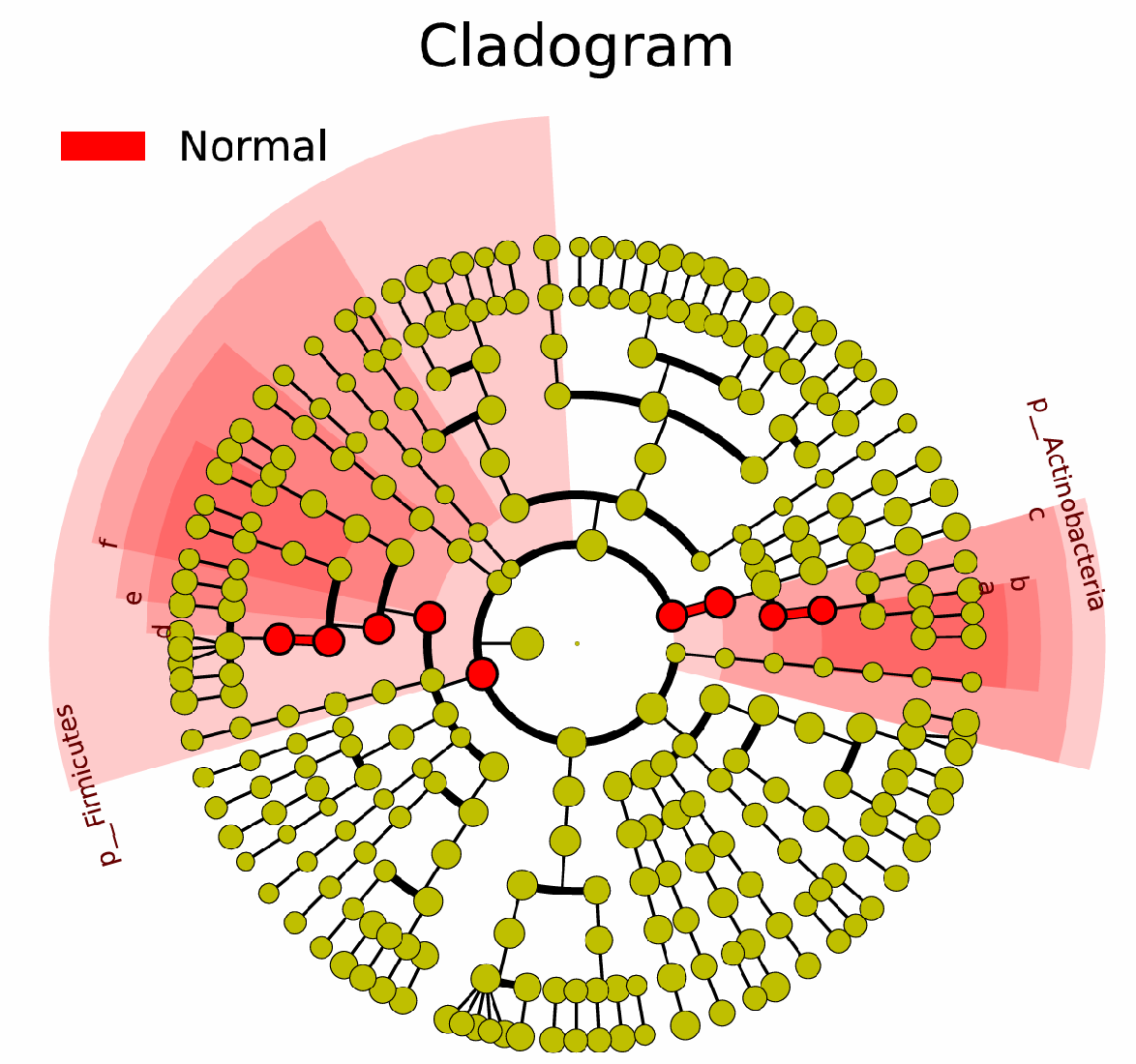
使用Adobe Illustrator进行图片拼接。其中,第1-6层(界、门、纲、目、科、属)的分类学注释在图片内进行,第七级(种级)在图片左上角用字母进行标注。
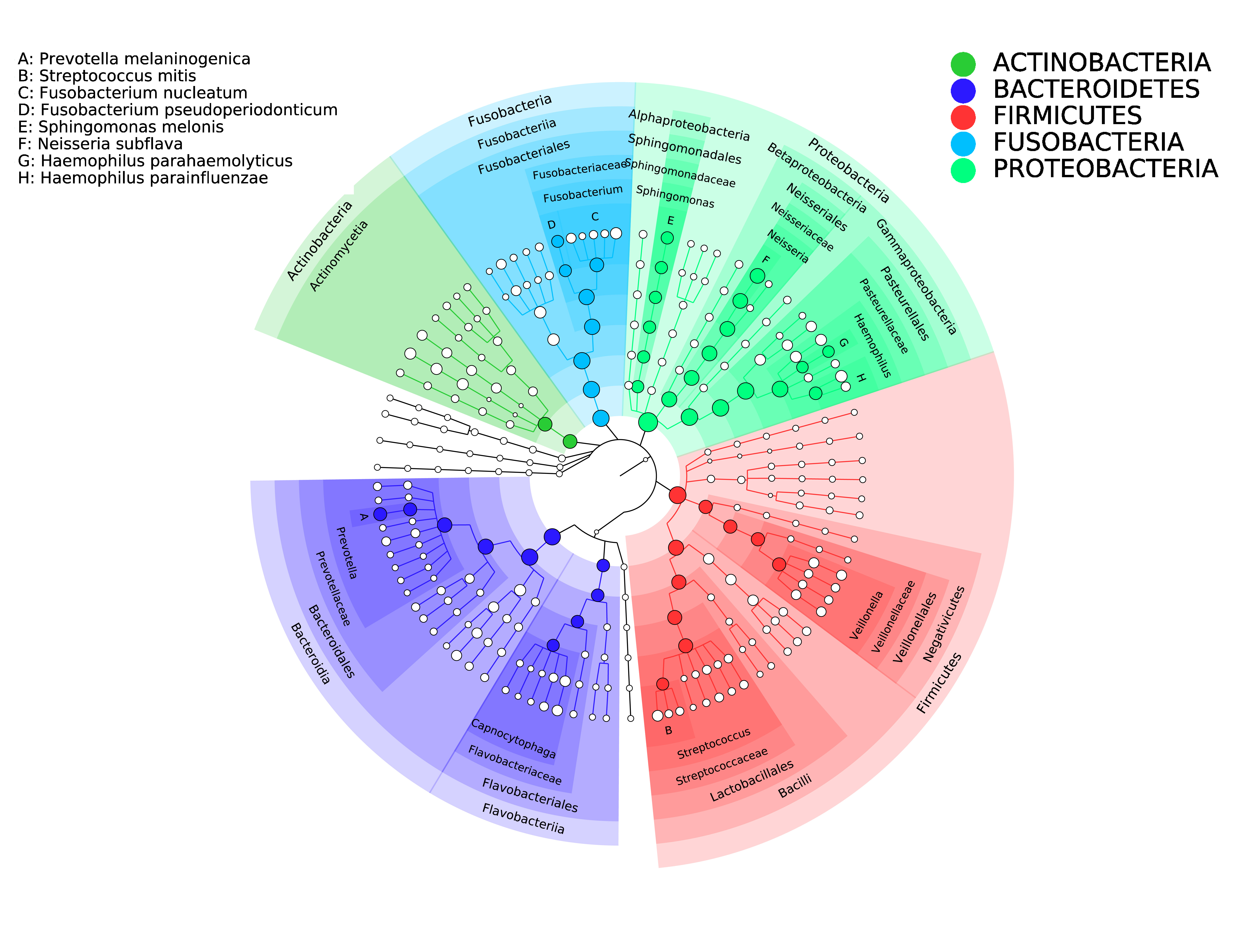
绘图方式2
R脚本graphlan_plot.r是新开发的物种树绘制脚本,提供了一种新型的绘图风格。其中关键的参数如下:
-n:设置显示的分类单元数目阈值(按丰度排序)。
-d:设置metadata的存放位置。
-c:选择分组依据(metadata的列名)。
-g:指定要分析的组别名称,用逗号隔开。如果全部分析则用all表示
-t:选择绘图种类,包括和heatmap、bar。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
|
cd result
# 下载tsv2spf.py,并使用该python脚本将tsv文件转化为可供绘图的*.spf文件。
wget -c https://github.com/joybio/graphlan_plot/raw/refs/heads/main/scripts/tsv2spf.py
chmod a+x tsv2spf.py
python ./tsv2spf.py -i metaphlan4/taxonomy.tsv -o metaphlan4/taxonomy_plot.spf
# 【关键步骤!】下载R脚本。这里R脚本依赖R包:optparse,在上文中已经下载过。可以使用"conda install r-optparse"下载
wget -c https://github.com/joybio/graphlan_plot/raw/refs/heads/main/graphlan_plot.r
# 边缘以热图进行展示
Rscript ./graphlan_plot.r \
-i metaphlan4/taxonomy_plot.spf\
-d metaphlan4/metadata.txt\
-c Group\
-g all\
-n 100\
-t heatmap\
-o metaphlan4/Heat_Results
# 边缘以条形图进行展示
Rscript ./graphlan_plot.r \
-i metaphlan4/taxonomy_plot.spf\
-d metaphlan4/metadata.txt\
-c Group\
-g all\
-n 100\
-t bar\
-o metaphlan4/Bar_Results
cd ..
|
LEfSe分析
LEfSe(Linear discriminant analysis Effect Size,线性判别分析效果大小) 是一种用于宏基因组数据分析的生物统计学工具,旨在识别不同实验条件下(如疾病状态、环境差异)显著差异的微生物类群或功能基因,即生物标志物。LEfSe分析的步骤主要分为以下三步:
- 首先在多组样本中采用的非参数因子Kruskal-Wallis秩和检验检测不同分组间丰度差异显著的物种;
- 再利用Wilcoxon秩和检验检查在显著差异物种类中的所有亚种比较是否都趋同于同一分类级别;
- 最后用线性判别分析(LDA)对数据进行降维和评估差异显著的物种的影响力(即LDA score)。
安装LEfSe
1
2
3
|
conda creat -n Lefse
conda activate Lefse
conda install lefse
|
处理原始数据
将数据处理成以下格式。其中第一行为分组信息,第二行为样本编号,之后的内容和taxonomy.tsv文件一致。
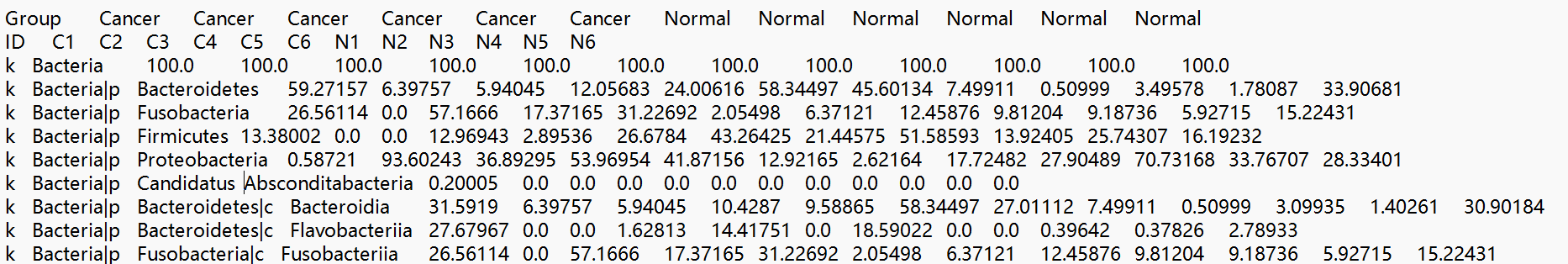
然后使用lefse_format_input.py将lefse.txt转化成程序的内部格式(非人类可读的形式)。其中有一些可选的参数:
-c:指定分组的大类在哪一行。这里分类信息在第一行。
-s:指定次级分组的信息在哪一行。这里没有次级分组,故填-1。(不能不填)
-u:指定样本名称的所在行。这里是第二行。
-o:缩放因子。LEfSe会对所有样本计算相对丰度后, 乘以这个缩放因子。缩放因子会影响LEfSe结果, 缩放因子越大, 倾向于获得更大的LDA值。通常固定为1000000, 以保证LDA的跨研究可比性, 否则, 通常的LDA筛选标准(LDA>2), 可能不适用于本次分析结果, 只能在本次分析结果内, 对feature的重要性排序。
1
2
3
4
5
6
7
8
9
10
11
12
|
# 提取样本行替换为每个样本一行,修改ID为SampleID
head -n1 result/metaphlan4/taxonomy.tsv|\
tr '\t' '\n'|\
sed '1 s/ID/SampleID/' >temp/sampleid
# 提取SampleID对应的分组Group(假设为metadata.txt中第二列$2),替换换行\n为制表符\t,再把行末制表符\t替换回换行
awk 'BEGIN{OFS=FS="\t"}NR==FNR{a[$1]=$2}NR>FNR{print a[$1]}' result/metaphlan4/metadata.txt temp/sampleid|tr '\n' '\t'|sed 's/\t$/\n/' >groupid
# 合并分组和数据(替换表头)
sed -n '1p' result/metaphlan4/taxonomy.tsv > sampleid
cat groupid sampleid <(tail -n+2 result/metaphlan4/taxonomy.tsv) >result/metaphlan4/lefse.txt
# 格式转换为lefse内部格式
lefse_format_input.py result/metaphlan4/lefse.txt \
temp/input.in -c 1 -s -1 -u 2 -o 1000000
|
进行Lefse分析
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
|
# 运行Lefse分析程序,输出结果为input.res
lefse_run.py temp/input.in temp/input.res
# 绘制物种数,标注差异
lefse_plot_cladogram.py temp/input.res \
result/metaphlan4/lefse_cladogram.pdf --format pdf
# 绘制所有差异features的柱状图
lefse_plot_res.py temp/input.res \
result/metaphlan4/lefse_res.pdf --format pdf
# 指定一个feature,绘制组间对比的图
lefse_plot_features.py -f one --format pdf \
--feature_name "k__Bacteria.p__Firmicutes" \
temp/input.in temp/input.res \
result/metaphlan4/lefse_Firmicutes.pdf
|


物种注释和丰度估计
通过Kraken2完成reads层面的物种注释和定量
创建Kraken2虚拟环境
1
2
3
4
5
6
7
8
9
|
conda create -c bioconda -n kraken2 kraken2
conda activate kraken2
# 创建数据库文件夹
DBNAME=databases/kraken2
mkdir -p $DBNAME
#直接下载标准数据库(约92.3GB)下载完解压至上述文件夹内
wget -c https://genome-idx.s3.amazonaws.com/kraken/k2_standard_20241228.tar.gz
|
运行kraken2命令
1
2
3
4
5
6
7
8
9
10
11
12
13
|
# 该步骤需要将80Gb左右的hash表读进内存,故电脑内存小于该值的情况下必须要加上--memory-mapping参数将文件映射至虚拟内存中。(速度可能会减慢!)
for i in `tail -n+2 metadata.txt | cut -f1`; do
kraken2 --db ${DBNAME}\
--paired temp/hr/${i}_?.fastq \
--threads 8 --use-names --report-zero-counts \
--report temp/kraken2/${i}.report \
--output temp/kraken2/${i}.output\
--memory-mapping
done
for i in `tail -n+2 result/metadata.txt | cut -f1`; do
kreport2mpa.py -r temp/kraken2/${i}.report \
--display-header -o temp/kraken2/${i}.mpa; done
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
|
mkdir -p result/kraken2
# 输出结果行数相同,但不一定顺序一致,要重新排序
tail -n+2 metadata.txt | cut -f1 | rush -j 1 \
'tail -n+2 temp/kraken2/{1}.mpa |\
LC_ALL=C sort |\
cut -f 2 |\
sed "1 s/^/{1}\n/" > temp/kraken2/{1}_count '
# 提取第一样本品行名为表行名
header=`tail -n 1 metadata.txt | cut -f 1`
tail -n+2 temp/kraken2/${header}.mpa | LC_ALL=C sort | cut -f 1 | \
sed "1 s/^/Taxonomy\n/" > temp/kraken2/0header_count
head -n3 temp/kraken2/0header_count
# paste合并样本为表格
ls temp/kraken2/*count
paste temp/kraken2/*count > result/kraken2/tax_count.mpa
|
